
第1章
ヒトゲノム入門
1DNA,ゲノムとは何か
1-1 ゲノム情報の構成
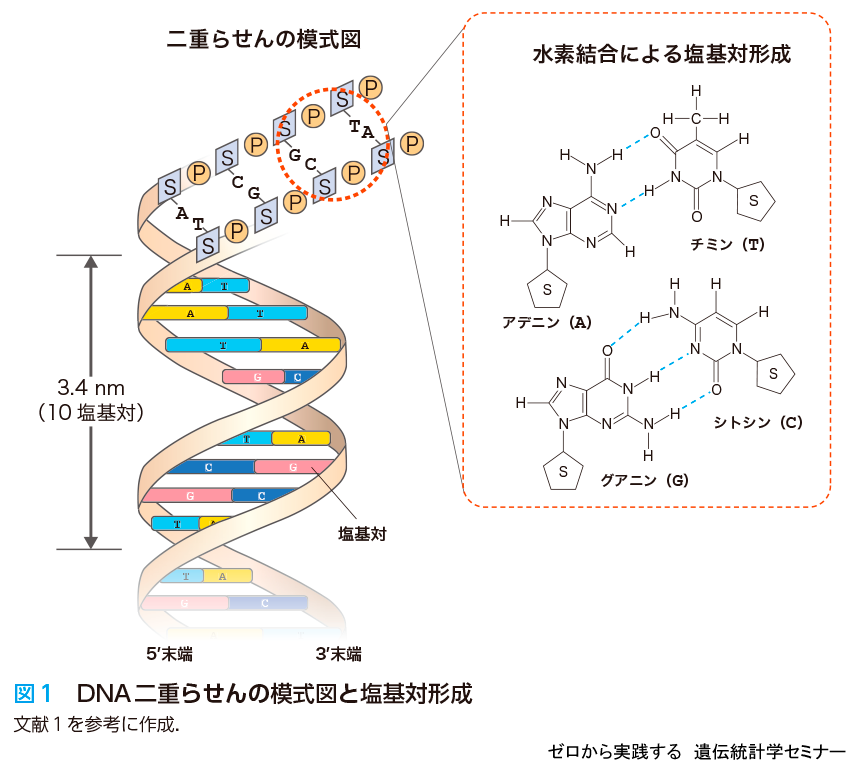
ゲノム情報はDNAに含まれるアデニン(A),チミン(T),グアニン(G),シトシン(C)の4種類の塩基で構成されています.「AとT」と「GとC」が水素結合を介してつながり,相補的な二本鎖を構成し二重らせん構造をとります.「GとC」は3本の水素結合でつながり,「AとT」は2本の水素結合でつながるため,「GとC」の結合力の方が強くなります(図1).
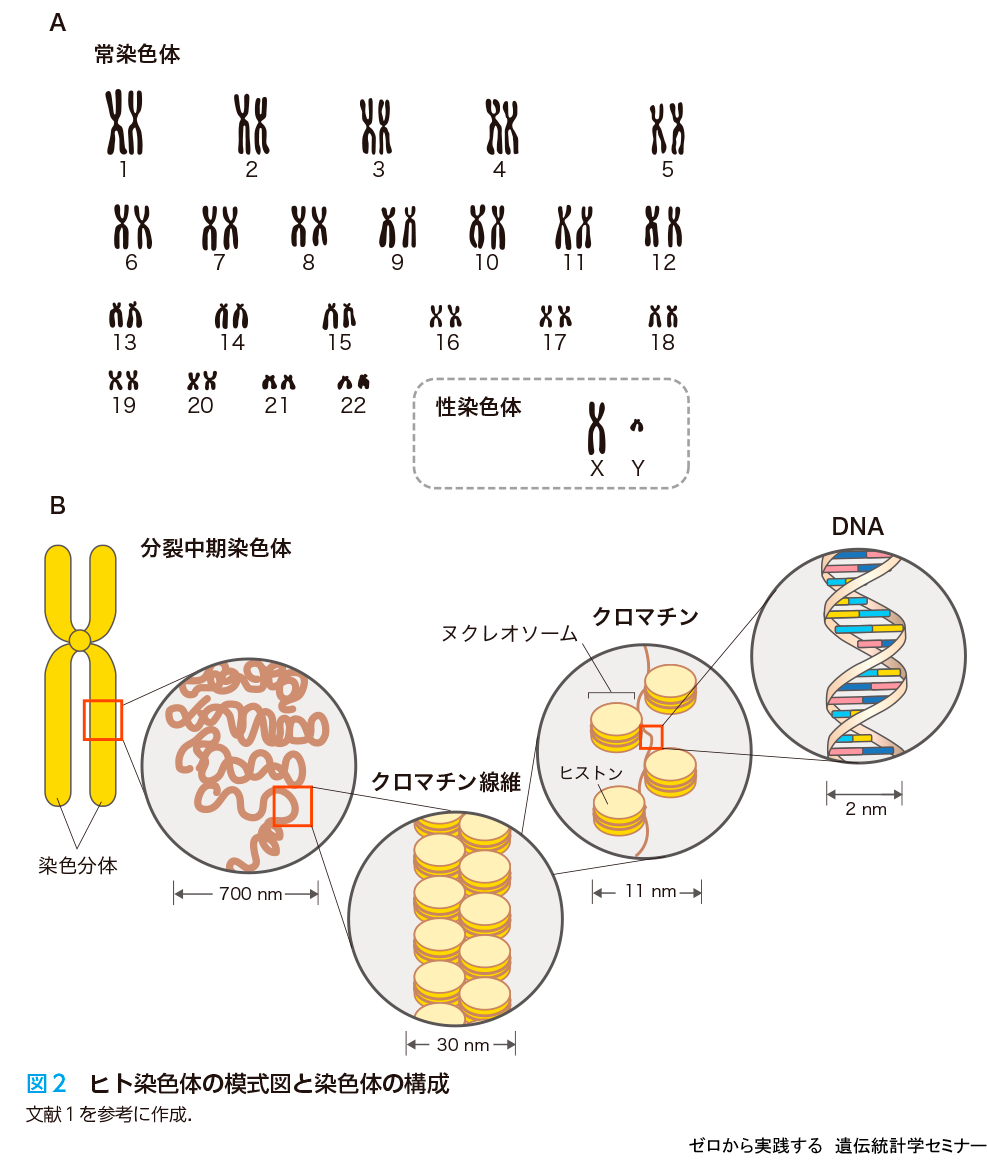
ヒトゲノム情報は46本の染色体にわかれています.ヒトは倍数体なので1番から22番までの常染色体を2本ずつもちます.これに加えて性染色体(X/Y)を2本もちます(図2A).この1番,2番…という染色体の番号は,染色体を顕微鏡で観察したときの長さが長い方から順に振られています.例えば染色体に含まれる塩基対の数は22番の方が21番より多いことが判明していますが,慣例がそのまま残っているために順番が逆転しています.各染色体の中には,図2Bに示すような階層でDNAが折り畳まれており,その上にはA,T,G,Cの4種類の塩基が配列しています.
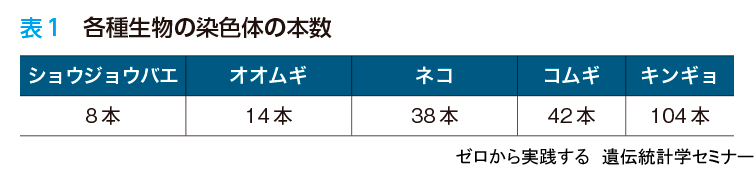
染色体数やゲノムの全長は,種によって大きく異なります(表1).以前は「染色体が多い」,「ゲノム配列が長い」といった種が進化的に高等であるといった考え方もありました.現在では,染色体数や染色体の長さは,高等であることの指標ではなく,各生物種にとって最適になるように決まっていることが分かっています.
ヒトの遺伝子数は?
ヒトゲノムが解読される前はヒト遺伝子の数が多く見積もられ,10万個ぐらいあると言われていました.2003年,ヒトゲノムが解読されたことで,現在ではヒトの遺伝子数は2万個ぐらいと言われています.
ところで,皆さんが実験をされていて,RNAやタンパク質を扱うことがあるかと思いますが,実験操作によってはこれらの物質はすみやかに失活してしまいます.一方,ゲノムDNAは非常に安定な物質であり,チューブに入れて室温で放置しても,数年経過しても実験に用いることができる場合もあります.このような安定性があるおかげで,例えば掘り出してきた古代人の骨からゲノムDNAを採取することも行えます.
ゲノムDNAは安定であると述べましたが,生物が生きていくなかで特に細胞分裂や,減数分裂の際に変異が入ることがあります.このような変異が子孫へと伝達され集団内に蓄積することで,個人間の違いや種の違いが生じます.例えば,ヒトの常染色体数は44本ですが,類人猿のものは46本です.ヒトと類人猿の共通祖先は46本の常染色体を持っており,ヒトに進化する過程で染色体の結合が起きて常染色体が1ペア減ったと考えられています.染色体同士の結合は変化の度合いが大きなイベントであるため異常も起こりやすく,融合した染色体をもつ個体は生存しにくい場合もありますが,ヒトへの進化の過程で起きた染色体の結合は比較的影響の小さい領域で起きたと想像できます.
1-2 ヒトゲノム
ヒトゲノムが約30億の塩基で構成されていることがわかったのは2000年代初頭のことでした.当時,ヒトゲノム配列全体の概要を解読するプロジェクトと並行して染色体1本の塩基配列の詳細な解読を複数の研究グループで分担して行っていました. 2001年にヒトゲノムのドラフト配列が報告され,その後2003年に精密配列が決定され,ヒトゲノムが30億の塩基配列より構成されることがわかりました2)3).かの有名なクレイグ・ヴェンター(J. Craig Venter)氏はこのヒトゲノム解読で活躍しました.
ここで示すゲノム解読とは,ごく少人数のゲノムDNAを元にしたヒトの標準的なゲノム配列の決定のことを指します.あくまで標準配列なので,皆さんそれぞれのゲノム配列とは少しずつ違います.標準配列を決定できた時点でゲノム研究は終わったと考える人もいましたが,各個人のゲノム間の差異の重要性が認識されはじめ,そちらの解析に研究者の関心は推移してきました.本書のメインテーマである個人差につながるわけです.
2ヒトゲノム配列とその個人差
2-1 個人差と一塩基多型(SNP)
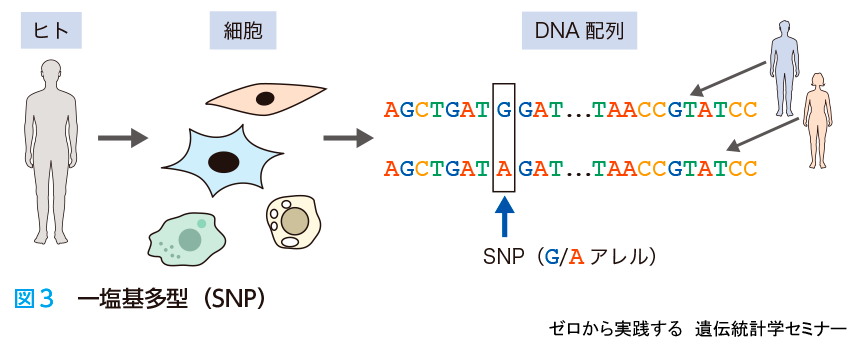
ヒトのゲノム配列は,個人間だけでなく,個人内でも少しずつ違います.個人間のゲノムの違いは多型(たけい)といい,英語ではpolymorphismといいます.多型にはいろいろな型があり,以前より知られているものとして繰り返し配列(リピート)が挙げられます.犯罪捜査で個人を決めるときに使われる,マイクロサテライトのくり返しの数に関する研究が,多型解析がはじまった頃にさかんに行われてきました.
その後,多型に関する知見が増えてきて,1塩基だけ違いのある多型であるSNP(single nucleotide polymorphism:スニップ)が個人差の決定に影響を与えていることがわかりました.図3の場合ですと,GがAになっているところがSNPです.SNPは比較的安定している変異です.図3のように一カ所だけGがAに変化してもゲノム全体として見たときにほとんど変化が起きておらず,安定的に子孫に受け継がれることが多いです.前述のくり返し配列は不安定なことが多いので,子孫に正確に伝わらないこともあります.したがって,最終的に個人のゲノムに起きた変異としてはSNPが多く受け継がれており,最近では個人差を調べる際にSNPを解析する傾向が高まっています.
2-2 ゲノムと個人差
ゲノムが違うと何が変わるのでしょうか? ここでは,外見の近さを例に挙げて説明します.ゲノムの近さは外見の近さで測ることができて,一卵性双生児はゲノム配列が同一であり,外見も瓜二つになります.次に近いのは兄弟,姉妹,親子であり,これらの関係では遺伝学的にゲノムが半分一緒といえます.半分一緒である場合,外見はある程度似通ってきます.ゲノムの違いが大きくなり,人種が変わるほどになると,髪の毛,皮膚の色,唇の形といった外見は大きく異なります.人種の違いの定義にはいろいろなものがありますが,ゲノムの観点からだとヒトゲノム配列が相対的に異なることと定義できます.
なぜ,これほどまでにも外見がゲノムで決まるのかを考察すると,外見は生物にとって大事な要素だからです.どういう形をしてるかというのは,外的環境への応答や運動能力も左右し,そのため生存に直結します.したがって,非常に選択圧を受けやすい要素であるため,生息地での生存に適した外見をもつ個体のゲノムがより多く残るようになります.
外見以外にも,生死にかかわる形質はゲノムと非常に強くかかわります.現代に生きる私たちにとっては外見が寿命に直結することはありませんが,その代わりに疾患の発症のしやすさ,薬剤の効きやすさが生死にかかわります.これらの形質は外見と同じようにゲノム配列で決定されることがわかっているため,ゲノム情報を医療の研究に活用するモチベーションへとつながります.
2-3 ゲノムの違いを解析した国際プロジェクト
■ 国際HapMap Project
ヒトゲノムの違いを解析する研究の集大成の一つが国際HapMap Projectです(図4).これはヒトゲノム全体を網羅するSNPのカタログをつくることを目的とした,国際コンソーシアムでした.このプロジェクトにかかわっていた研究者達は欧米,アジア,アフリカから集めた270名のSNPの内容を解読しました.当時はまだ商用のSNPマイクロアレイは実用化されていなかったので,染色体ごとに解析を担当する国や研究室を割り振って,ひたすらサンガー法でシークエンスを行い,少しでも塩基が標準配列と異なる箇所があったら塩基変異のパターンを調べていくことが手作業で進められました.このプロジェクトにかかわっていた研究室には,シークエンサーがたくさん並んでいて,それらを24時間体制で動かし続けて解析していたようです.このプロジェクトのおかげで,2000年代前半に200万~300万のSNPが明らかになり,以前は一生懸命手探りで遺伝子クローニングしていたものを,網羅的に解析できるようになりました.
ゲノム配列の決定からSNP同定への推移
私が学生であった2000年代は“クローニング”という言葉を多く聞きました.クローニングとは,生化学的な精製で見つかったタンパク質を対象として,そのタンパク質をコードする遺伝子を決定する作業を指します.タンパク質からアミノ酸配列は比較的簡単に決められますが,そのアミノ酸配列を決める塩基配列のパターンには数多くの可能性が考えられるため,さまざまな塩基配列のパターンを試して,ゲノムのどの領域に該当する遺伝子があるかを縮重PCR(degenerate PCR)とよばれる方法で探し出していました
1980〜’90年代はクローニング競争とよばれ,いかに他の研究者より先に重要な遺伝子をクローニングに成功するかを競い合っていました.ヒトゲノムの配列が決定され,SNPに関する知見が増えてくると研究の方向性が変わって,ゲノム配列の地図をもとに個人差を決めるSNPを同定する研究もさかんになりました.
このプロジェクトは国際プロジェクトでしたが,日本語のウェブサイトもありました.じつは日本がこのプロジェクトにおいて一番貢献していたというのをご存知でしょうか.SNP解析対象者の270名の中に,45名の日本人のサンプルが入っていました.日本人のSNPデータがこのプロジェクトによって公開されたため,その後のゲノム研究では強力なアドバンテージを得ることができました.
私が初期研修医であった2005年頃にこの国際HapMap Projectに関する『Nature』の記事4)を読みましたが,それは私にとって衝撃的なものでした.当時は大学院進学を希望して研究室を探していたところで,このプロジェクトのウェブサイトを早速閲覧しました.これまでのクローニングによるアプローチとは大きく異なり,全体的なゲノム情報が多数集まった状態から研究を網羅的に進めていく方向性がこのプロジェクトを皮切りにはじまったと感じました.この出来事がきっかけとなりゲノム解析を自分の研究のメインテーマに据えようと思いました.

■ 1000 Genomes Project
国際HapMap Project以降もヒトゲノムを解読するプロジェクトは続きました.国際プロジェクトとしては,「1000 Genomes Project」が有名です(図5).プロジェクト名のとおり1,000人のゲノムを解読するプロジェクトであり,最終的には約2,500名分のゲノムが解読されました5).次世代シークエンサー(next generation sequencer:NGS)を使って網羅的に解析が行われ,だいたい1億カ所のSNPが見つかりました.ヒトゲノム配列の長さは30億塩基ですから,30塩基ごとに1塩基ぐらいは世界中の誰かが変異をもっているといったイメージになります.世界中のあらゆる人種のゲノムを解読したプロジェクトであり,現生人類がもっているゲノム配列の大半が明らかになりました.
2-4 ゲノム配列の解読手法とゲノム情報の活用に関する変化
■ゲノム配列の解読手法の推移
ゲノムを読む方法には大きく分けて2つあります.1つはサンガー法です.サンガー法は,塩基配列を決定する方法のうち,一番正確な配列を得られる方法といえます.いろいろな変異解析手法がありますが,本当に新しい変異であるかを確認する際には,現在もこの手法を用います.サンガー法の欠点として,時間や費用が結構かかる点と,PCRの鋳型として比較的大量のゲノムを消費してしまう点があげられます.
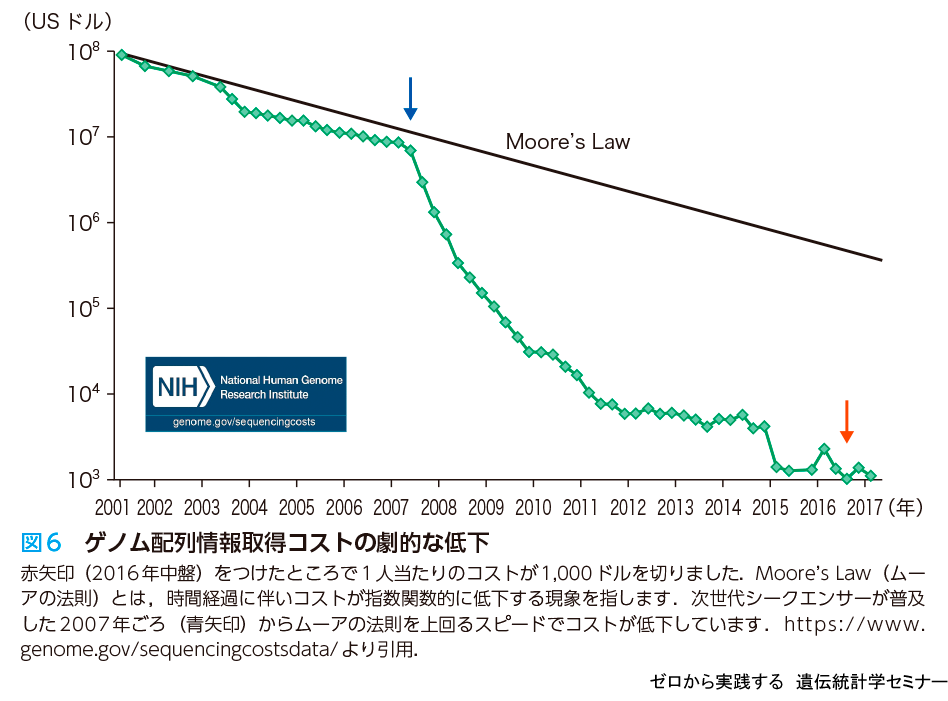
最近では次世代シークエンサーを用いてゲノム解析を行うことが多くなりました.ゲノム解析がはじまった2000年頃は,全ゲノム配列を読むのに1人当たり1億ドルかかっていましたが,その後の次世代シークエンサーの開発もあり現在では1人当たり1,000ドルを切りました(図6).私の研究室でも全ゲノム解読のコストは外注で1人7万円程度になっており,非常に安く・早く解読できる時代になりました.
また,コストが低下しただけでなく,全ゲノムシークエンスの精度が高くなり,解析パイプラインの改良により,シークエンスデータの信頼性も高くなってきました.
■ マイクロアレイ解読コストの低下
SNPマイクロアレイで解読する費用はさらに安くなっています.最近の50万〜60万SNP解析用のSNPマイクロアレイでは,1サンプル5,000円ほどで行えます.これはゲノムDNA抽出費用とほぼ同じ値段になります.また,このSNPマイクロアレイの精度はかなり高く,サンガー法とほとんど変わりません.このようにゲノム解析は,以前と比べてはるかに手軽に行えるようになってきました.
現在私の研究室では,DNAやRNAの抽出までは行いますが,その後のシークエンス実験については外注先の企業や他の研究室に依頼する方法をとっています.
■ ヒトゲノム研究の大規模化
解析コストの大幅な低下に伴って,研究の大規模化が進んでいます.例えば,英国のUKバイオバンク※16)は50万人の全ゲノムSNPデータ,TOPMedプロジェクト※2は10万人の全ゲノムシークエンスデータを構築済みです(図7).また,UKバイオバンクは,ゲノム・表現型情報を安価で分譲しています.

■ ゲノム研究の商業化
次世代シークエンサーを,研究施設だけでなく企業が保有する例が増えています.年々更新される高額機器を頻繁に購入し,購入金額に見合うだけの運用を続けるのは,研究施設より企業の方が有利となりつつあります.また,研究者が企業にヒトゲノムの解読を外注する仕組みができました.大容量のデータが得られるため,得られたデータを研究に使う際にはスーパーコンピューターを用いて解析しますが,Amazonなどの大企業が保有するクラウドサーバーの一部をレンタルして行う例が増えています.
2-5 Precision Medicineが宣言された後の変化
オバマ前米大統領が2015年に一般教書演説で宣言した“Precision Medicine”という言葉には強いインパクトがありました.端的に表すと「ゲノム情報を医療に活用しましょう」ということです.
以前は,ゲノム情報は研究者が自分の研究のために集めていた側面が強く,患者さんに得られた知見を還元する段階までなかなか届いていませんでした.ところが,この言葉が発せられてからは,患者さんのゲノム情報を持ち主の患者さんに還元するといった,至極当たり前のことが当たり前の目的として掲げられるようになりました.日本におけるゲノムに関連した研究の風向きについても,世の中でゲノム情報を活用しようといった機運が高まってきているように感じています.
疾患ゲノム解析では必ず誰かにサンプルをいただいています.サンプルをくれた患者さんは,病態を解明してほしい,薬を創って欲しいと思ってサンプルを提供してくださいます.したがって,ゲノム解析をするうえで,このことを心に留めおいて研究を進める必要があります.
summary
- ゲノム情報の構成と一塩基多型(SNP)の違いによる個人差について概要を説明しました.
- 2003年にヒトゲノム標準配列が決定されました.
- ヒトゲノム解読後に国際HapMap Projectや1000 Genomes ProjectなどのSNP解析の国際プロジェクトが行われました.
- ヒトゲノム,SNPマイクロアレイの解析コストが劇的に低下し,大規模なゲノムデータ解読が進みました.
文献
- 「診療・研究にダイレクトにつながる遺伝医学」(渡邉 淳/著),羊土社,2017
- Venter JC, et al:Science, 291:1304-1351, 2001
- International Human Genome Sequencing Consortium:Nature, 409:860-921, 2001
- International HapMap Consortium:Nature, 426:789-796, 2003
- 1000 Genomes Project Consortium:Nature, 526:68-74, 2015
- Bycroft C, et al:Nature, 562:203-209, 2018
- 『マンガでわかるゲノム医学 ゲノムって何?を知って健康と医療に役立てる!』(水島-菅野純子),羊土社,2018