
第1章 マイクロバイオーム研究をはじめる前に考えること
1 ヒトマイクロバイオーム研究の全体像
服部正平
本稿ではこの20年間におけるヒト常在微生物叢(ヒトマイクロバイオーム)研究の推移を紹介しつつ,サンプル調製やシークエンス技術,各種マイクロバイオームデータの取得,被験者の宿主・生活様式データの収集,これらの異層データ間の相関・因果関係の解析など,マイクロバイオーム研究における解析手法の現状とそこに含まれる問題点やピットフォールを解説する.
1マイクロバイオーム研究の始まりと推移
今日の国際的なヒトマイクロバイオーム研究が始まったのは,世界初のホールゲノムショットガンによるヒト腸内マイクロバイオームのメタゲノム解析論文の発表や国際ヒトマイクロバイオームコンソーシアム(IHMC)が設立された2006〜2008年である1)〜3).この時期は,ヒトゲノム解読に代表される大量の遺伝子・ゲノム情報(=DNA塩基配列データ)を基盤としたデータ駆動型研究の有効性が生命科学分野で広く認識された頃であった.このような背景のなか,多くが難培養性で未知微生物種からなるヒトマイクロバイオーム研究はメタゲノム解析というデータ駆動型アプローチとして開始された.
この概ね20年間のヒトマイクロバイオーム(腸内,口腔,皮膚)研究の変遷をPubMed論文数の推移で調べると,2006〜2024年のヒト腸内マイクロバイオームの論文の累積数は約52,000報で,2024年だけで約9,000報であった(図1).3つの生息部位別の論文数の割合(2015〜2024年の10年間)は腸内:口腔:皮膚=76:17:7であった.この10年間のヒト腸内マイクロバイオームの論文数の平均前年比は1.36で(COVID-19の影響による2022年と2023年での減少がみられる),この値は他の研究テーマ(例えば,human pathogen genomeの論文数の10年間の平均前年比は約1.08)よりも有意に高い増加率を示す.一方,2006〜2024年の日本からのヒト腸内マイクロバイオームの論文の累積数は約2,800報で,2024年だけで約400報であった.この値から,日本からの論文数の割合は世界全体の5%程度と推定される(図2).3つの生息部位の論文数の平均比(2015〜2024年の10年間)は腸内:口腔:皮膚=71:24:5となった.10年間(2015〜2024年)の腸内マイクロバイオームの論文数の平均前年比は約1.45で,世界と同じく高い増加率を示した(同様にCOVID-19の影響による2022年と2023年の減少がみられる).これらの現状から,ヒトマイクロバイオーム研究は現在も発展しており,長期の国際的な重要テーマと考えられる.以上の論文数のサーベイは十分に精査していないため必ずしも正確とは言えないが,世界と日本のざっくりとした動向を示していると考えられる.
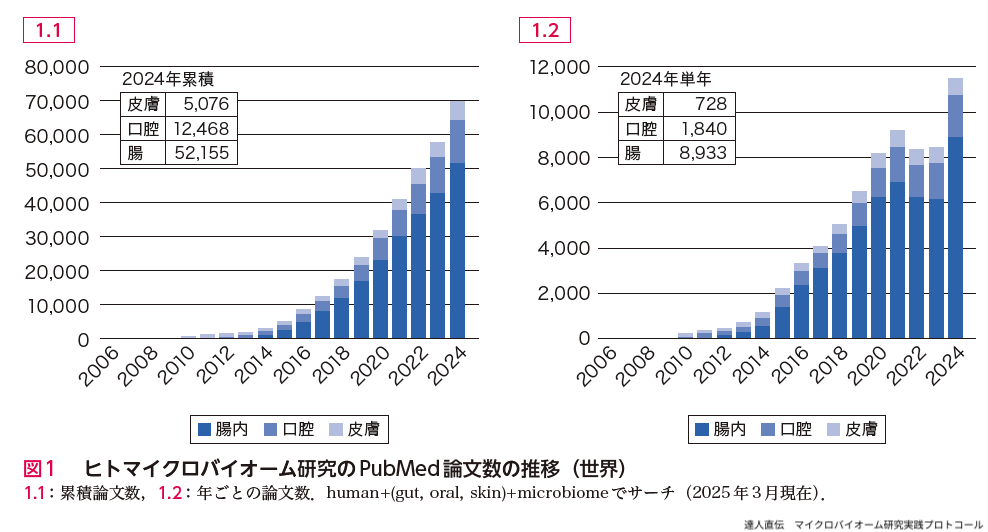
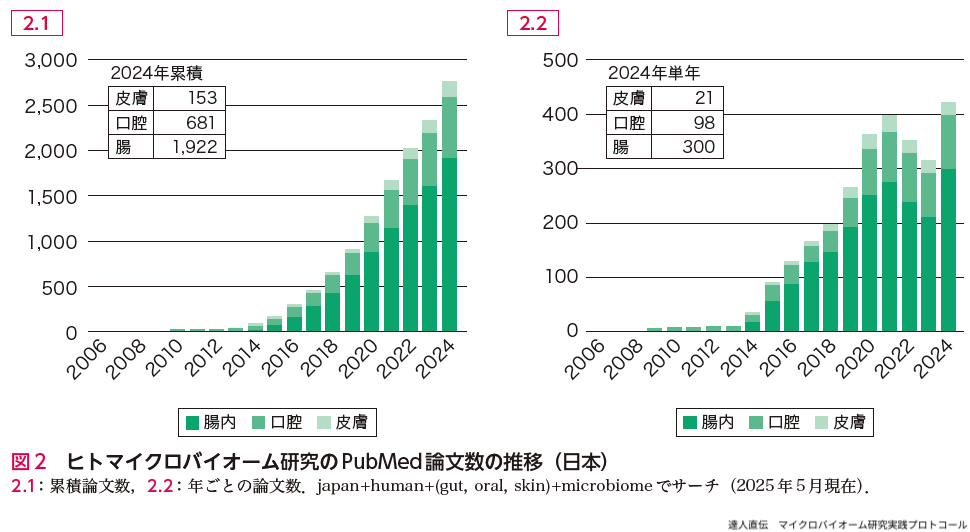
この20年間に蓄積されてきた論文のなかには,今日のヒトマイクロバイオーム研究に多大なインパクトを与えた注目すべき論文がいくつかある.すべてを網羅することはできないが,「微生物種の系統解析法の開発」,「肥満の人の糞便が肥満の発症に寄与する」,「健常者の便微生物移植が腸炎患者を治療する」,「腸内短鎖脂肪酸が宿主の恒常性維持に働く」,「エンテロタイプの発見」,「高い個人間多様性と生息部位特異性」,「ヒト遺伝子をはるかに凌駕した遺伝子数と多様性」,「免疫細胞の分化誘導能をもつ腸内微生物種の存在」,「環境因子と腸内マイクロバイオーム多様性の強い関係性」などをあげることができる4)〜15).これらの論文は,ヒトマイクロバイオーム研究において解析技術の発展が重要であり,またヒトの健康/病気と密接な関係があるなど,ヒトマイクロバイオーム研究を推進する科学的意義を明確化した.
2マイクロバイオーム研究で収集するデータ
今日のヒトマイクロバイオーム研究の中心は,次世代シークエンス(NGS)技術による高速シークエンシングと高速・高性能コンピューティングを原動力とする疾患患者のマイクロバイオームとその比較対照である健常者のマイクロバイオームを対象としたデータ駆動型研究といえる.疾患マイクロバイオーム研究のめざすところは疾患発症メカニズムの解明であり,健康マイクロバイオーム研究は健康(恒常性)維持メカニズムの解明である.これらのメカニズムに関与する微生物種やその産物,宿主遺伝子などの探索は,(個別化)精密医療とヘルスケア(疾患の診断・治療・予防)の実現化につながる16)〜18).
今日のヒトマイクロバイオーム研究で収集されるデータを図3にリストする.そのリソースは非侵襲性の糞便,唾液,皮膚などの生体試料であり,研究目的によっては生検粘膜や手術摘出組織,羊水,痰などもリソースとなる.マイクロバイオームのDNA配列を収集する第一ステップは生体試料からのDNA抽出である(2章-1,2章-2).ついで,NGS技術を用いて,抽出したDNAから細菌・アーキア(2章-2,2章-3,3章-1,3章-2),酵母などの真核微生物(2章-6),バクテリオファージ(2章-4,3章-3)に由来する塩基配列データがメタゲノムデータとして収集され,そのアセンブリからはmetagenome assembled genome(MAG)と総称される個々の微生物種ゲノムのコンティグが再構築される.特殊なケースとして,遺跡から発掘された古代人の骨や歯などもメタゲノムデータ収集のリソースとなり,例えば,歯石のメタゲノムシークエンスは,その時代の口腔微生物種や食習慣などの情報を探索する手段となる(3章-4).また,生体試料から抽出したRNAからは遺伝子転写物の塩基配列データであるメタトランスクリプトームデータが収集される.

初期のマイクロバイオーム研究は遺伝子探索に重きをおいたホールゲノムショットガンによるメタゲノム解析(以下,メタゲノム解析)が切り拓いたが,コスト面や迅速性などの理由で,今日では微生物種探索を主眼にした16S遺伝子解析による研究が多数を占めている.一方で,同じ種だが宿主への生理作用や機能が異なる菌株が存在すること,健常者と疾患患者から分離された同種異株が異なったゲノム構造/遺伝子をもつことなどが明らかとなり19)〜21),さらには最近の微生物ゲノムデータベースの拡充もあいまって,種組成や種数の定量評価が不確実な16S遺伝子データよりも,全ゲノム配列やハウスキーピング遺伝子セット(mOTU)の類似度を指標にしたより精密な株(ゲノム)レベルでの種特定と種数・種組成解析が汎用傾向にある22)23).実際,高インパクトファクタージャーナルに掲載される論文のほとんどはメタゲノムデータを用いている.よって,今後のシークエンスコストの低減(サンプルあたりのデータ量の増加)を期待すると,近い将来には16S遺伝子解析は予備的解析となり,メタゲノム解析が主流になると考えられる.
DNA・RNAデータのほかに,質量分析機(MS)や核磁気共鳴装置(NMR)を用いて,生体試料から調製された低分子量の化合物分画からは分子構造(と分子量)でデータ化された微生物の代謝産物(メタボローム)データ(2章-5),生体試料から調製されたタンパク質画分からはアミノ酸配列でデータ化されたメタプロテオームデータを収集することも一般化している.収集されたDNA,RNA,代謝物,タンパク質データの総体データ(オミクスデータ)を統合したマルチオミクス解析が今日のヒトマイクロバイオーム研究のトレンドとなっている.特に糞便や血中のメタボロームはヒトマイクロバイオームの機能実体あるいはマーカーとして重要視されている.
くわえて,ヒトマイクロバイオーム研究(と微生物学全般の研究)の推進に不可欠の基盤技術・研究として個々の微生物株の分離培養・保存や系統分類がある.例えば,特定の分離培養株は宿主―微生物間相互作用を解明する生物実験(ノトバイオートマウスの作製など)に有効利用される(1章-3,4章-1).また,個々の分離培養株のゲノムデータは,MAGと合わせて微生物ゲノムデータベースの拡充を促進し,その日々のアップデートは,現在だけでなく将来におけるマイクロバイオーム研究(とすべての生命科学研究)の発展を担保するライフラインと位置付けられる(1章-2).
さらに,今日のマイクロバイオーム研究では,宿主データ(年齢や性,BMIなど)や生活様式データ(食事,飲酒や喫煙,運動,疾病・投薬歴など),ヒトゲノムデータ(SNVsやメチル化など)の収集は必須項目となっている.これらの被験者関連データはそれらとマイクロバイオームデータとの関連解析(5章-1,5章-2)に活用され,ヒトマイクロバイオーム研究の主要テーマである宿主・生活様式―マイクロバイオーム―宿主表現型(遺伝子)の関係性の解明にリンクする(後述).
3ヒトマイクロバイオーム研究のデータ解析とピットフォール
1 交絡因子
ヒトマイクロバイオーム研究は,宿主・生活様式―マイクロバイオーム―宿主表現型(宿主遺伝子)の3因子間の相関・因果関係の解明に収斂すると考えられる(図4).つまり,これまでの疫学研究で示唆されている宿主・生活様式―宿主表現型の関係(図4c)へのマイクロバイオームの関与(媒介)とそのメカニズムを,宿主・生活様式―マイクロバイオーム間の関係(図4a)とマイクロバイオーム―宿主表現型間の関係(図4b)から解き明かすアプローチである.特に,疾患腸内マイクロバイオーム研究(図4b)において疾患患者群の腸内マイクロバイオームの変容(dysbiosis)はさまざまな疾患で観察されており,これらの疾患への特定の微生物種の関与が示唆されている.しかし,観察されたdysbiosisには宿主・生活様式の影響(図4a)が多分に含まれるので,被験者全員の宿主・生活様式データの収集は必須項目となる.腸内マイクロバイオームに影響する宿主・生活様式因子には,年齢,性,BMI,食事,投薬,飲酒,ブリストールスケール(便形状スケール)などがあり9)10)24)〜27),これらはマイクロバイオーム―宿主表現型間の関係(図4b)の正確な評価における交絡因子(疾患とマイクロバイオーム間の関係性を撹乱する因子)となる.よって,健常者群と疾患患者群(特に,疾患患者群では投薬の影響がないナイーブ患者)間の宿主・生活様式データに統計的有意性がない群の設定,信頼度の高い統計検定に十分な被験者数(サンプル数)の設定とそれから逆算した(数倍の?)リクルートすべき被験者数の算出,2群間のマイクロバイオームを区別する最大の因子がその疾患であることの確認など,用意周到な研究計画が望まれる.逆に言えば,これらの交絡因子を考慮していない,あるいは不十分な論文にはミスリーディングが多分に含まれる可能性があり,それを見極めるには論文中の交絡因子に関する記載を注意深くチェックする必要がある.このような交絡因子の調整は主に健康な被験者を対象とした食材/食品の介入試験(介入群とプラセボ群の2群間比較)における腸内マイクロバイオームの解析にも必須である.特に,交絡因子の収集と調整が不十分にならざるを得ない被験者数の少ない介入試験では,元来その生理作用が小さい食材/食品の効果よりも未収集あるいは未調整のより強い交絡因子の効果が大きく反映されたアウトカムになる可能性を否定できず,よく検討された研究計画とアウトカムの慎重な解釈が望まれる.
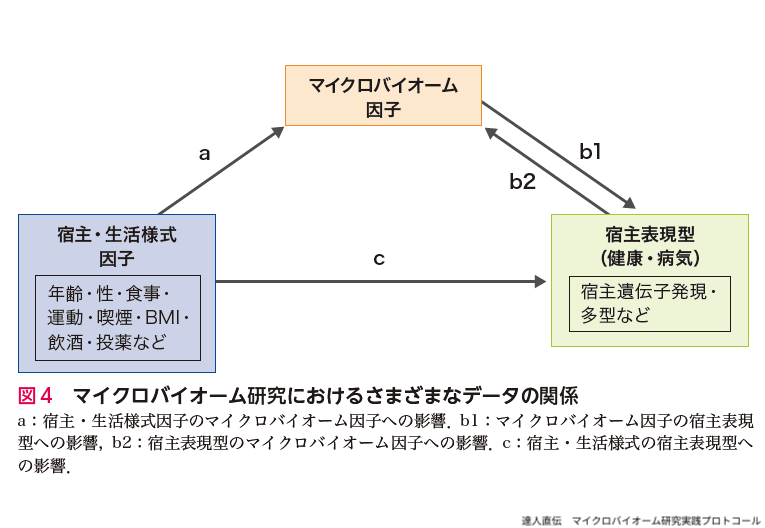
上記は被験者の宿主・生活様式にかかわる交絡因子だが,最近の研究から実験系のDNA抽出法がヒト腸内マイクロバイオームの多様性に影響する交絡因子となることが報告された28).この研究は公的データベースに登録されている世界中の16S配列データ(168,000サンプル)のバラツキに関与する因子を調べ,16S遺伝子可変V領域の違い,DNA抽出法の違い,国・地域の違いがサンプル間のバラツキを説明する因子として特定された.16S遺伝子可変領域(V1〜V9)における異なった16S遺伝子可変V領域から得られた菌種組成の間には互換性がないことは以前から知られており,この結果は当然といえる.また,国・地域間の高い多様性についてもすでにわれわれがメタゲノムデータを用いて報告している29).この国・地域の特異性は固有であるため,異なった国・地域のデータを集積した多コホート研究(いわゆるメタアナリシス)よりも同一国・地域の被験者からなる単一コホート研究のアウトカムの方がより有効であることを示唆する.注目すべきはDNA抽出法の違いが腸内マイクロバイオームのバラツキに大きく影響することである.DNA抽出法については本書の2章-1と2章-2に詳しいが,ウェット実験のなかでDNA抽出法の違いが他のサンプル保存やシークエンス工程の違いよりも腸内マイクロバイオームのバラツキに大きく影響することがすでに指摘されている30).つまり,例えば,日本の異なったグループが異なったDNA抽出法を用いて得た同じ疾患のマイクロバイオームデータを比較した場合,用いたDNA抽出法間のマイクロバイオームのバラツキの程度が疾患―健康間のマイクロバイオームのバラツキの程度よりも有意に小さければ解析結果は有効であり,大きければ解析結果は無効となる.
2 因果関係
マイクロバイオーム―宿主表現型間の関係(図4b)の解明において,観察されたdysbiosisが疾患発症の原因(図4b1)なのか結果(図4b2)なのかはこの段階では判断できない.宿主の生理状態の変化に同調するマイクロバイオームの変化(図4b2)の例として,腸内や唾液マイクロバイオームで観察される24時間周期の菌種組成の概日リズムをあげることができる31)32).よって,この因果関係をさらに調べる必要があり,それには無菌マウスに疾患患者の糞便(腸内マイクロバイオーム)あるいは疾患と相関する(複数の)微生物種を移植して作製したヒト化ノトバイオートマウスの表現型を調べる方法がある.しかし,このノトバイオートマウスを用いる方法には,対象のヒト疾患と全く同じ疾患がマウスで起こるのか,疾患と相関する微生物種の生理作用(炎症など)が対象のヒト疾患の発症と直接リンクするのかなど,実験デザインそのものの妥当性や結果の解釈などに対する問題提起がある33).
マイクロバイオームと疾患の因果関係のもうひとつの例として,多くの疾患マイクロバイオームの特徴として観察される短鎖脂肪酸(酢酸や酪酸)産生微生物種の有意な減少をあげることができる.この現象は発症メカニズムが異なる疾患で共通することから,疾患の原因というよりも疾患の結果(図4b2)と考えるのが合理的である.このことを逆説的に原因微生物種が存在すると仮定すれば,短鎖脂肪酸の減少を伴う原因微生物種(図4b1)はいまだ特定されていないことになる.一方,原因微生物種は存在しない(つまり,結果である)と仮定すれば,宿主・生活様式因子にその疾患の直接の原因が含まれると推定される.
3 その他
大腸がん組織へのFusobacteriumの高頻度検出など,微生物の他臓器へのトランスロケーションが参考となり20)34),微生物が通常存在しないと考えられている臓器・組織などにマイクロバイオームの存在を調べる16S遺伝子解析が増えてきている.しかし,このようなごく微量のマイクロバイオーム解析では,周辺環境の微生物の夾雑(コンタミ)やサンプル間のクロスコンタミは必然であり,コンタミに起因した間違った結果や解釈にしばしばつながる.例えば,胎児マイクロバイオームの研究で,その存在を肯定する結果と否定する結果が複数のグループから発表された.両グループともコンタミを考慮したネガコン実験をしていたが,この正反対の結論に至った理由は,肯定派は検出された微生物種をコンタミではないと判断し,存在を否定したグループは検出された微生物種をコンタミと判断したという解釈の違いであった.結局,第三者グループが両グループのデータを再検討し,胎児にはマイクロバイオームは存在しないという結論となった35).
同様に,皮膚マイクロバイオームもかなり微量であるため36),通常の実験操作ではコンタミは免れない.よって,ネガコン実験でのコンタミ微生物種のリストアップは最低限必要である37).くわえて,皮膚マイクロバイオームの16S遺伝子解析に関して,もうひとつ注意すべき点がある.われわれも体験しているが,皮膚の優勢菌種であるCutibacterium acnesは可変領域V1-V2で検出できるが,可変領域V3-V4およびV4はプライマー配列のミスマッチのため,ほぼ検出されないかV1-V2に比べてその存在比は著しく低下する38).このことは2016年に発表されているにもかかわらず,V3-V4を用いた皮膚マイクロバイオームの論文が今でも散見される.
おわりに
ヒトの健康/病気には遺伝(先天的)要因と環境(後天的)要因の2つが関与するといわれて久しい.遺伝要因の実体であるヒトゲノムが2000年初めに全解読され,これにより遺伝子というデジタル情報(転写量など)を用いて遺伝要因を定量評価することが可能となった.一方,マイクロバイオームの知見が蓄積されるにしたがって,マイクロバイオームが環境要因を反映したデジタル情報となることがわかってきた.つまり,さまざまな生活様式を含む環境要因(アナログ情報)の変化や違いを定量可能なマイクロバイオームデータ(デジタル情報)の変化や違いに変換でき,ここに,ヒトの健康/病気に関与する遺伝要因と環境要因をデジタル情報として科学的に評価・理解できる時代が来たといえる.
文献
- Gill SR, et al:Science, 312:1355-1359, doi:10.1126/science.1124234(2006)
- Kurokawa K, et al:DNA Res, 14:169-181, doi:10.1093/dnares/dsm018(2007)
- Mullard A:Nature, 453:578-580, doi:10.1038/453578a(2008)
- Lozupone C & Knight R:Appl Environ Microbiol, 71:8228-8235, doi:10.1128/AEM.71.12.8228-8235.2005(2005)
- Turnbaugh PJ, et al:Nature, 457:480-484, doi:10.1038/nature07540(2009)
- Caporaso JG, et al:Nat Methods, 7:335-336, doi:10.1038/nmeth.f.303(2010)
- Fukuda S, et al:Nature, 469:543-547, doi:10.1038/nature09646(2011)
- Arumugam M, et al:Nature, 473:174-180, doi:10.1038/nature09944(2011)
- Human Microbiome Project Consortium:Nature, 486:207-214, doi:10.1038/nature11234(2012)
- Yatsunenko T, et al:Nature, 486:222-227, doi:10.1038/nature11053(2012)
- van Nood E, et al:N Engl J Med, 368:407-415, doi:10.1056/NEJMoa1205037(2013)
- Atarashi K, et al:Nature, 500:232-236, doi:10.1038/nature12331(2013)
- Li J, et al:Nat Biotechnol, 32:834-841, doi:10.1038/nbt.2942(2014)
- Atarashi K, et al:Cell, 163:367-380, doi:10.1016/j.cell.2015.08.058(2015)
- Rothschild D, et al:Nature, 555:210-215, doi:10.1038/nature25973(2018)
- Schüssler-Fiorenza Rose SM, et al:Nat Med, 25:792-804, doi:10.1038/s41591-019-0414-6(2019)
- Gilbert JA, et al:Nat Med, 31:1099-1113, doi:10.1038/s41591-025-03615-9(2025)
- Hibberd MC, et al:Nature, 625:157-165, doi:10.1038/s41586-023-06838-3(2024)
- Atarashi K, et al:Science, 358:359-365, doi:10.1126/science.aan4526(2017)
- Nakamoto N, et al:Nat Microbiol, 4:492-503, doi:10.1038/s41564-018-0333-1(2019)
- Nii T, et al:Ann Rheum Dis, 82:621-629, doi:10.1136/ard-2022-222881(2023)
- Parks DH, et al:Nucleic Acids Res, 50:D785-D794, doi:10.1093/nar/gkab776(2022)
- Milanese A, et al:Nat Commun, 10:1014, doi:10.1038/s41467-019-08844-4(2019)
- Nagata N, et al:Gastroenterology, 163:1038-1052, doi:10.1053/j.gastro.2022.06.070(2022)
- Nishijima S, et al:Cell, 188:222-236.e15, doi:10.1016/j.cell.2024.10.022(2025)
- Vujkovic-Cvijin I, et al:Nature, 587:448-454, doi:10.1038/s41586-020-2881-9(2020)
- Vandeputte D, et al:Gut, 65:57-62, doi:10.1136/gutjnl-2015-309618(2016)
- Abdill RJ, et al:Cell, 188:1100-1118.e17, doi:10.1016/j.cell.2024.12.017(2025)
- Nishijima S, et al:DNA Res, 23:125-133, doi:10.1093/dnares/dsw002(2016)
- Costea PI, et al:Nat Biotechnol, 35:1069-1076, doi:10.1038/nbt.3960(2017)
- Thaiss CA, et al:Cell, 167:1495-1510.e12, doi:10.1016/j.cell.2016.11.003(2016)
- Takayasu L, et al:DNA Res, 24:261-270, doi:10.1093/dnares/dsx001(2017)
- Walter J, et al:Cell, 180:221-232, doi:10.1016/j.cell.2019.12.025(2020)
- Zepeda-Rivera M, et al:Nature, 628:424-432, doi:10.1038/s41586-024-07182-w(2024)
- Kennedy KM, et al:Nature, 613:639-649, doi:10.1038/s41586-022-05546-8(2023)
- Sender R, et al:PLoS Biol, 14:e1002533, doi:10.1371/journal.pbio.1002533(2016)
- Kurokawa R, et al:Sci Rep, 13:19666, doi:10.1038/s41598-023-46890-7(2023)
- Gohl DM, et al:Nat Biotechnol, 34:942-949, doi:10.1038/nbt.3601(2016)