
第1章 基本フローと各フローにおける検討ポイント
1. CRISPRスクリーニングで最も重要な「数」を理解する
遊佐宏介,樽本雄介
CRISPRスクリーニングの実験フローは複数に分割することができるが,各ステップにおいてノイズやサンプリングエラーを低減させることが検出感度向上に重要である.特にサンプリングエラーを理解することは,なぜ大規模な実験スケールが必要なのかを考えるうえでポイントである.これを理解することで「プロトコール通りの細胞数」ではなく,各自の実験系にカスタマイズした実験規模を設定することが可能となるだろう.複雑な表現型アッセイをCRISPRスクリーニングに適用する場合には,特によく理解して系の構築を進める.
はじめに
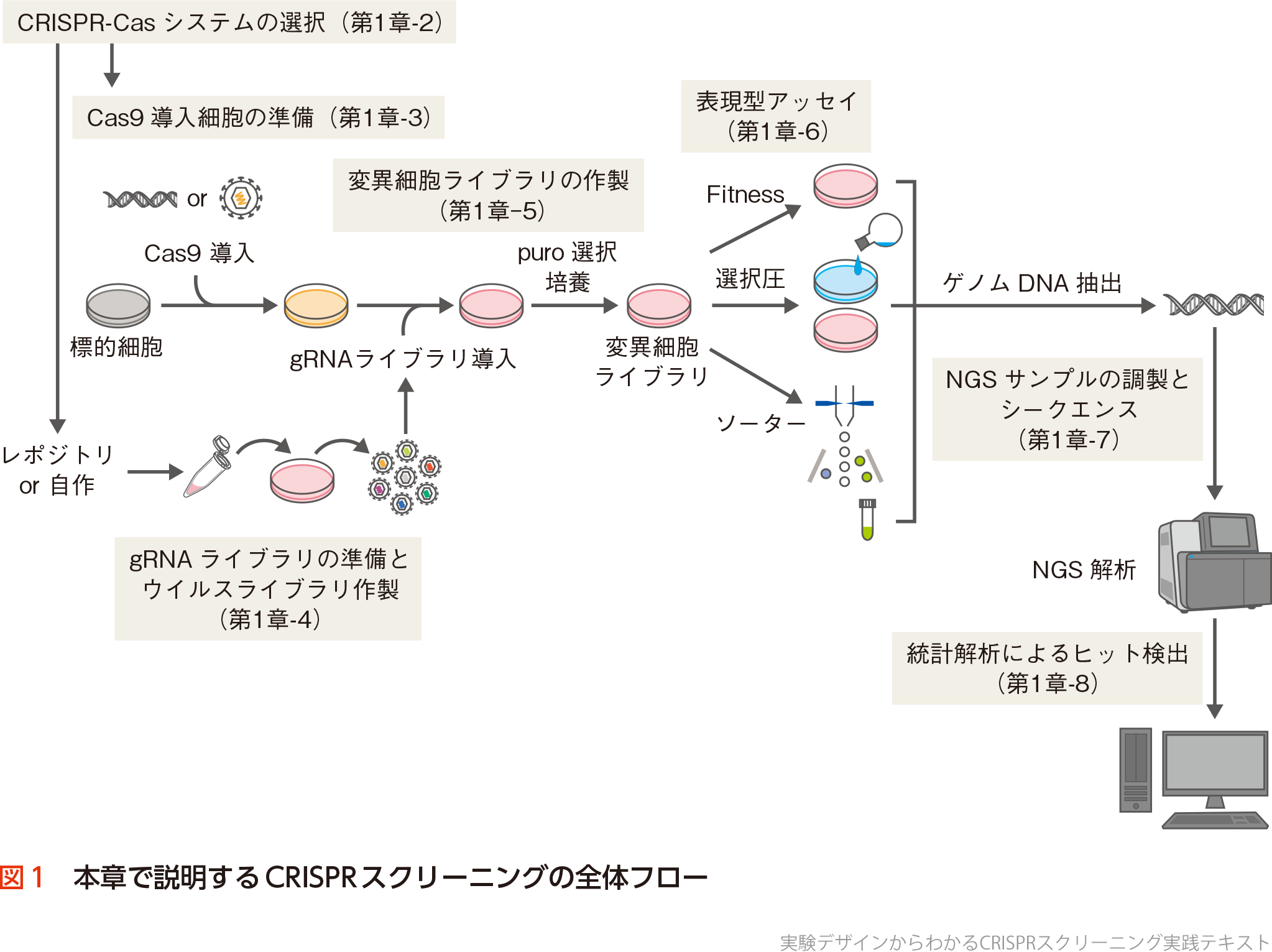
実験材料の準備も含めたCRISPRスクリーニングの全体フローは図1のようになる.これを7分割し,第1章で基礎知識,重要ポイントやノウハウを解説する.CRISPRスクリーニングは,タンパク質をコードする約2万遺伝子すべてに対し,遺伝子あたり4~6個のgRNAを設計し,合計8~12万gRNAを1つにまとめた(プールした)状態で扱うプール型スクリーニングが一般的である.第1章では計算を単純化するために,総遺伝子数2万,遺伝子あたり5 gRNAからなる合計10万(100k) gRNAのゲノムワイドライブラリを考える.
一連の流れのなかで,3つの「ライブラリ」が登場する.①gRNAライブラリ,②変異細胞ライブラリ,③NGSライブラリである.CRISPRスクリーニングの場合,gRNAはレンチウイルスベクターに搭載し発現させるため,gRNAライブラリはプラスミドDNAとレンチウイルスの2形態をとる.さらに,クローニング前のオリゴヌクレオチドも含めると3形態ともいえる.どのライブラリを指しているのかに注意してほしい.
本稿では,まず,多くのステップに関係し,必ず理解しておく必要がある「数」の基本を解説し,ノイズの発生源や100k gRNAを一度に扱う実験とはどのようなスケールなのか理解を深める.
complexityとcoverageとポアソン分布
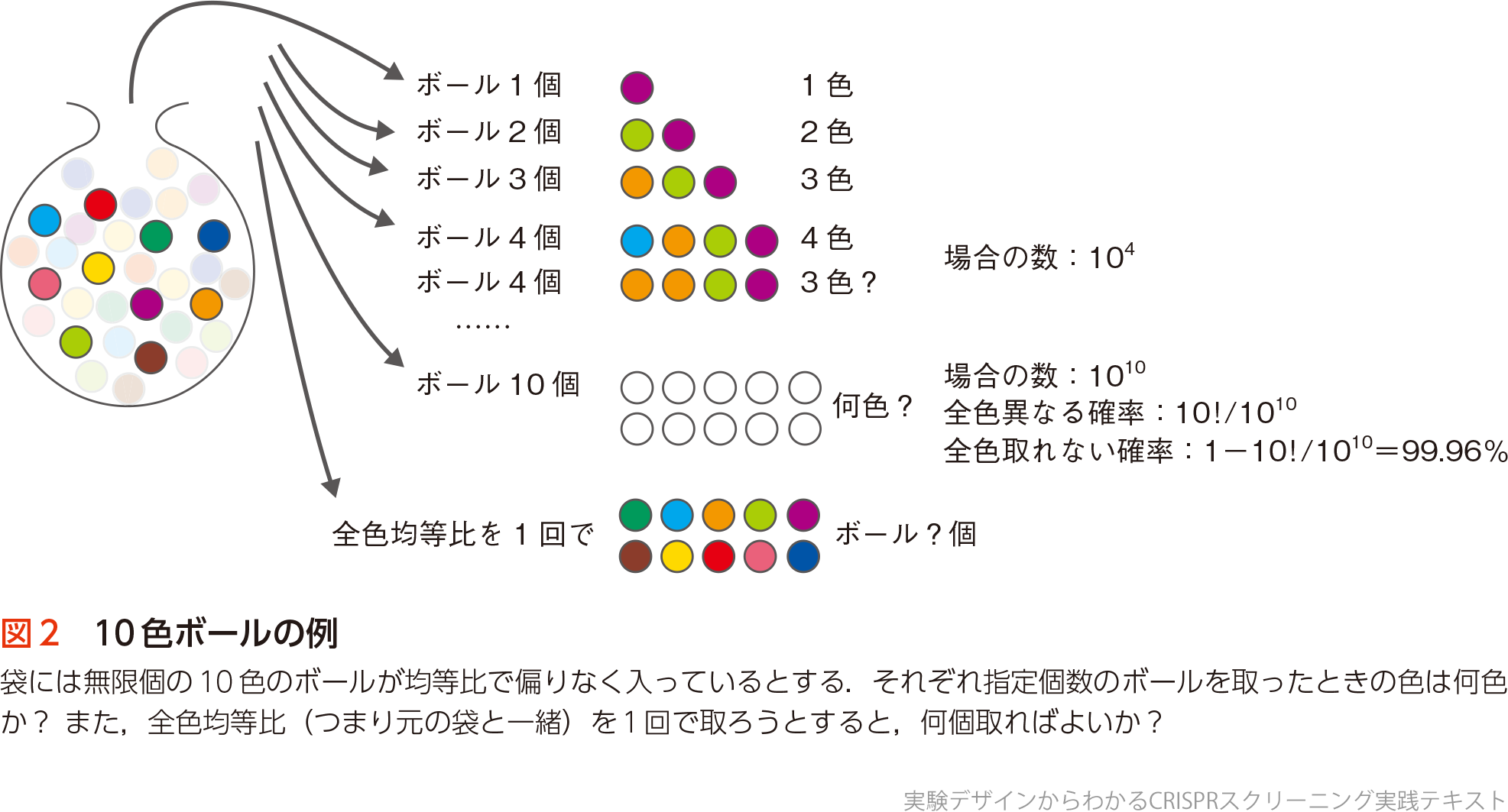
まず,クイズから……10色のボールが同じ比率で,またよく混ざった状態でたくさん入っている袋がある(図2).このなかからある個数のボールを1回取り出した場合,何色のボールを取ることができるか?…… 1個なら当然1色.2個なら2色.3個ならまあ3色? 4個で4色だが,同じ色を引いてしまうと3色…….10個取って全色引き当てる確率は,10!/1010=3.6×10−4.簡単に場合の数や確率が計算できるだろうが,ボールを単純に10個取っただけでは,まず間違いなく(99.96%)失う色があることは想像に難くないだろう.つまり,10色の集まりという多様性(complexity)を失う.では,元の比率(1:1:1……)と同じ比率で10色を取るには何個取ればよいのか(全部はなし)? 言い換えると,母集団の多様性を維持した子集団をつくるには,どのくらいの規模のサンプリングを行う必要があるだろうか?
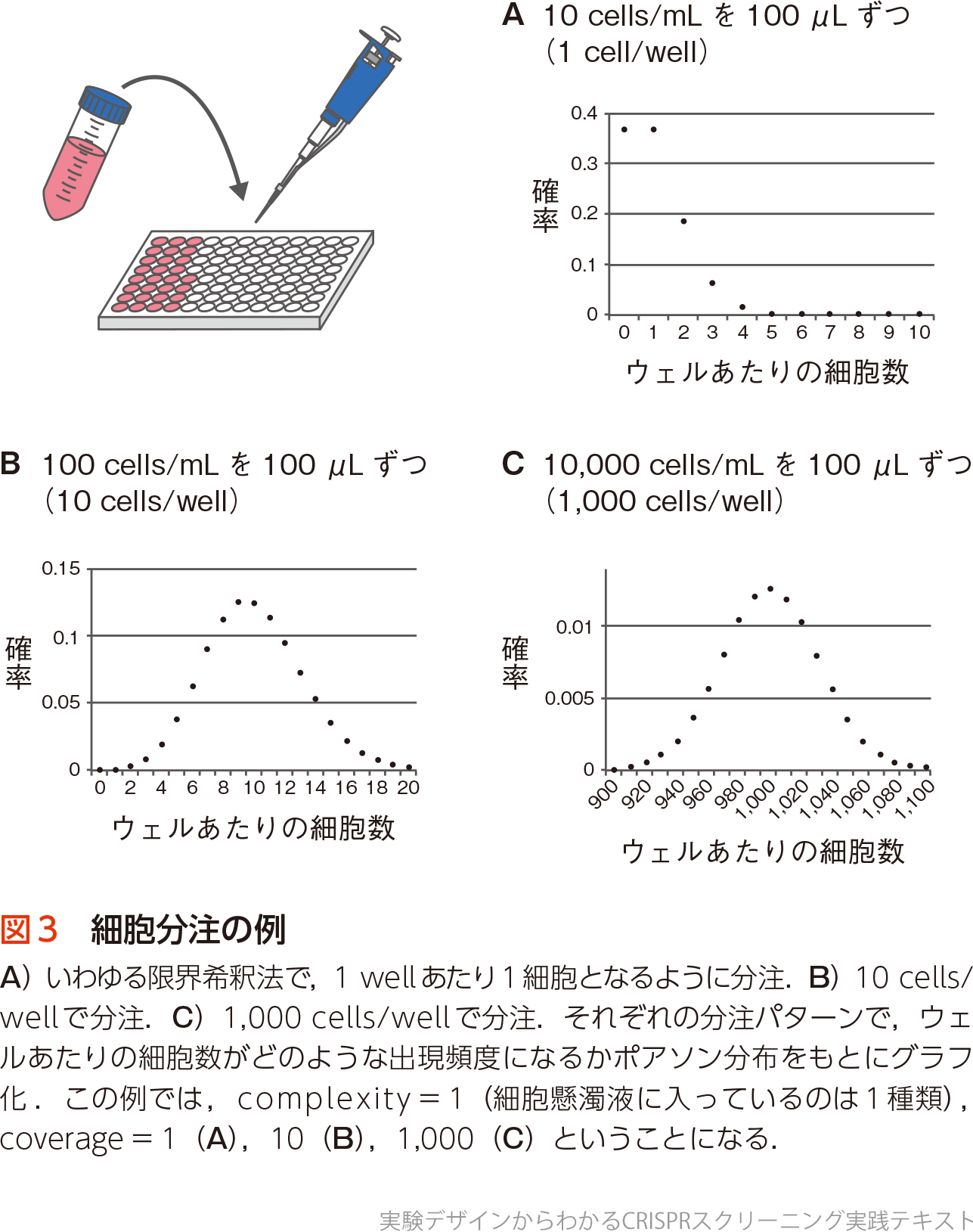
もう1つ,生物学実験を行う研究者に身近な例を考えてみたい.限界希釈による細胞のクローニング実験である.細胞懸濁液を10 cells/mLに希釈し,96ウェルプレートに100μL/wellで細胞を蒔きこむ(つまりlimiting dilution法.single-cell sortingではない).平均すると各ウェルに1細胞が蒔かれたこととなり,細胞が増えてくれば1細胞由来クローン(single cell-derived clones)を取得できるはずだが,現実は?
この実験例やボールの例ではポアソン分布を使えばその答えにたどり着くことができる(コラム参照).限界希釈の場合,実際にはすべてのウェルに1細胞ずつ入ることはない.空ウェル,1細胞ウェル,2細胞ウェル,それ以上の細胞数が蒔かれたウェルが存在し,きちんと実験できていれば1細胞のウェル数は35くらいになる(図3A).1細胞クローニングの実際の成功率は播種直後の細胞の生存,またその後の増殖の結果によるので単純に35ではないだろう.なぜ,1細胞入ったウェル数が35なのか? それはいくら1 cell/100μLの細胞懸濁液とはいっても,毎回100μLのピペッティング操作で必ず1細胞を分取できるわけではないからである.細胞を顕微鏡下で取ったり,セルソーターで分ける等すれば別であるが,よく混和した細胞懸濁液から100μLピペッティングする場合,分取できる細胞数の分布はポアソン分布に従い,1 cellから外れた値,つまり平均値からのずれ,サンプリングエラー(取れなかったり,多めに取れたり)が発生する.これは避けることができない.
次に,細胞濃度を10倍上げて100 cells/mLの細胞懸濁液を100μLずつ分注しよう.10 cells/100μL/wellでの分注だが,ポアソン分布を見てみるとおおよそ3〜18 cells/wellの間となり,さすがに空ウェルはほぼないが10細胞ウェルは10%程度しかない(図3B).つまり,正確に元の細胞濃度と同じ濃度で分取できるのは10回に1回で,あとはすべて濃度が変わり,大まかに0.3〜1.8倍の間となる.このぶれがサンプリングエラーで,母集団と子集団で濃度を変えてしまう.
では,一気に細胞濃度を上げて10,000 cells/mLとして,100μLずつ分注する.1,000 cells/wellでの分注となり,分布を見ると900〜1,100の間に収まり,元の細胞濃度からのぶれは大きくても±10%までに縮小していることがわかる(図3C).10%もぶれてしまう確率は非常に小さく,どのウェルを見ても1,000 cellsに近い数で分注される.つまり,サンプリングエラーは小さくなり,ピペッティング操作ごとのばらつきも抑えられたことになる.
上記の限界希釈の例では細胞は単一の細胞種と仮定していたが,では,複数の細胞株の混合液ではどうだろうか? 仮に5種類の細胞株が均等に混合された細胞懸濁液を考えてみよう.1,000 cells/wellの分注でよいだろうか?…… 答えは×.1,000 cells/wellで分注しても,細胞株ごとに見ると200 cells/wellで分注されていることとなり,1細胞種2,000 cells/mLを100μL分注しているのと同じことになり,それぞれの細胞種にサンプリングエラーが発生する.細胞総数を見るとばらつきは少ない(毎回1,000 cellsくらい取れている)が,その内訳は分注ごとにばらついている.というわけで,それぞれの細胞株につき1,000 cells/wellとなるように,5種類あるので,5,000 cells/wellでの分注が正解である.
この細胞分注例で言うところの,細胞株数5が混ざり物(ライブラリ)の多様性(complexity)であり,分注時の各細胞株の細胞数1,000がカバレッジ(coverage)となる.complexity×coverageが分注する細胞総数となる.
では,10色のボールの入った袋から何個取り出せば,母集団の均等比を子集団で再現できるか.答えから言うと比は多少変わってしまうのだが,10,000個(各色あたり1,000個)取り出すと,比の変化がきわめて微小に抑えられた子集団をつくることができる.
CRISPRスクリーニングにおけるサンプリングエラー
上記の例で,母集団から一部を分取(サンプリング)するような操作を行う場合,母集団の多様性とカバレッジを考慮しないと,母集団とは多少異なる子集団となることがわかったと思う.この「ばらつき」がサンプリングエラーであり,ノイズとなる.
では,実際にCRISPRスクリーニングでは,いつ,分取=サンプリングという操作を行っているのか?…… 母集団としてはgRNAライブラリのプラスミドDNAを考える.正確にはスターティングマテリアルはオリゴプールであるが,実質gRNAライブラリプラスミドが母集団でよい.
分取を行うステップは,次のようになる.
❶ gRNAライブラリプラスミドの一部をトランスフェクションに使い,レンチウイルスを作製する.
❷ 作製されたレンチウイルスの一部が,細胞に感染する.
❸ 感染細胞の一部を使い,継代する.
❹ 増えた細胞の一部を使い,さらに継代をくり返す.
❺ 増えた細胞の一部に選択圧を加える,あるいはソーターで一部を取り出す.
❻ ゲノムDNAを鋳型にPCRを行い,gRNA部分を増幅し,NGSライブラリを作製
する.
❼ 作製したNGSライブラリをシークエンスする.
ざっと見てもこれらの操作……つまりすべて(!)の操作がサンプリング操作である.細胞操作だけを考えたらよいわけではない.実質気にしなくてよいのは❶のみである.❷〜❼の操作はすべて,細胞何個,DNA何g,リード数何カウントと分取を行っており,サンプリングエラーが入るステップである.前述のようにcoverageが低くなるとサンプリングエラーは大きくなる.いかにサンプリングエラーを抑えるか,入るとしても小さく抑えるか,許容範囲はどこかを考える必要がある.最終のNGSデータにはこれらのエラーが蓄積されて現れる.コントロールに対してサンプルに大きなノイズが入っていると,統計解析は不利になり,検出感度が下がり,ヒット遺伝子が取れなくなってしまう.もし遺伝子ヒットを見出せなかった場合,実験系の検証を行う必要がある.このとき,サンプリングエラーに起因するS/N比の低下が原因か,実際に遺伝子を取ることができなかったのかを判断する必要がある.サンプリングエラーの可能性を排除するためにも適切な実験操作を行い,「良い」データの取得が重要である.
プール型スクリーニングはRNA interferenceによるshort-hairpin RNAライブラリの時代から始まっており,そのときに良好なS/N比を確保するために1,000×カバレッジが目安となったようである.ゲノムワイドライブラリ100k gRNAsを使った場合,このカバレッジを達成するために扱う数はcomplexity(100,000)×coverage(1,000)=1×108となる.分取する際にはこの数を最低限とすることで,ばらつきが抑えられることを意味するが,単純にこの数字を上記の各操作に当てはめると以下のようになる.
❷ 作製されたレンチウイルスの一部を使い,細胞に感染(1×108感染細胞)させる.
❸ 感染細胞の一部(1×108細胞)を使い,継代する.
❹ 増えた細胞の一部(1×108細胞)を使い,さらに継代をくり返す.
❺ 増えた細胞の一部(1×108細胞)に選択圧を加える,あるいはソーターで一部(1×108細胞)を取り出す.
❻ スクリーニング後に細胞(1×108細胞)からゲノムDNAを抽出する.
❼ ゲノムDNA(1×108細胞)を鋳型にPCRを行い,gRNA部分を増幅し,NGSライブラリを作製する.
❽ 作製したNGSライブラリをシークエンスする(1×108リード).
上記の規模感はどうだろうか.実際にこの規模で実験を行うのは正直辛い.例えば,細胞継代を見ると,白血病細胞K562の推奨播種濃度は1×105 cells/mLで,1×108細胞を蒔きこむのに1,000 mLの培養液,T75フラスコ40個(25 mL/flask)またはT175フラスコ20個(50 mL/flask)が必要になる.接着細胞の場合,1×106 cells/10-cm dish播種なら,10-cm dish 100枚,15-cm dish約35枚となる.15-cm dish 35枚は,インキュベーターの半分ほどのスペースを占領する.ゲノムDNAの抽出はマニュアルで行えば扱う細胞数は問題にならないかもしれないが,ゲノムDNA抽出キットを使う場合,maxi-prepカラムの上限が1×108 cellsである.PCRに1×108 cells由来のゲノムDNA 660μgを鋳型にできるか? NGS解析でサンプルあたり1×108 reads取得は,RNA-seqでは5〜20×106 reads/sampleのデータを取得していることを考えると,シークエンスコストは膨大になる.
このように1,000×カバレッジは理想ではあるが,すべての操作において実現するのは無理があり,第1章の各項目で具体的に解説するように,適宜ダウンスケーリングして実験操作を行う.文献1にカバレッジを下げたときの影響をシミュレーションしているので参考にしてほしい.余裕のあるステップはカバレッジを高く,厳しいところは妥協してと,実際の操作に即して適宜カバレッジを調整し,それでも全体としてはノイズを許容範囲に抑えるよう実験を進める.
ご覧ください
文献
- Doench JG:Nat Rev Genet, 19:67-80, doi:10.1038/nrg.2017.97(2018)