概論
新しい疾患修飾薬,早期診断薬の開発に向けて 神経変性疾患の次の突破口
Next breakthrough toward developing novel disease modifying therapy and diagnostics against neurodegenerative disease
富田泰輔
Taisuke Tomita:Laboratory of Neuropathology and Neuroscience, Graduate School of Pharmaceutical Sciences, The University of Tokyo(東京大学大学院薬学系研究科機能病態学教室)
アルツハイマー病をはじめとする神経変性疾患は,タンパク質蓄積病態に伴って生じる「慢性代謝疾患」としての理解が深まりつつある.すなわち,細胞内外において病因タンパク質が構造転換し凝集していくプロセスと,この蓄積病態に対する脳内環境の恒常性維持機構ならびに炎症システムの変容が10年単位で継続した結果,最終的に神経細胞死そして疾病の発症に至ると考えられている.そういった発症プロセスを踏まえ,発症前診断につながるバイオマーカーの開発や新規創薬モダリティの導入が精力的に進められている.本特集においては新しいコンセプトに基づいて大きな展開を見せつつある国内外の神経変性疾患の基礎研究および創薬開発研究を紹介したい.
はじめに
アルツハイマー病(Alzheimer’s disease:AD),レビー小体型認知症(dementia with Lewy bodies:DLB),前頭側頭葉変性症(frontotemporal dementia:FTD)は,進行性の認知機能低下を呈する神経変性疾患であり,三大認知症ともよばれている.一方,黒質のドーパミン神経細胞の変性を主体とするパーキンソン病(Parkinson’s disease:PD)は振戦や固縮,無動を特徴とする.これらの神経変性疾患はいずれも主に中年~老年期に発症することから,超高齢社会を迎えたわが国において,大きな社会問題となっている.
いずれの疾患においても,著明な神経細胞死が認められるが,加えて,それぞれの疾患に特徴的な異常構造物が出現する.1906年にアルツハイマー教授によって報告された,ADにおける老人斑,神経原線維変化をはじめとして,それぞれの疾患の剖検脳において観察される異常構造物については多くの神経病理学者らによって精力的に解析,記述されてきた.しかしこれらの分子的な実態については長らく不明であった.
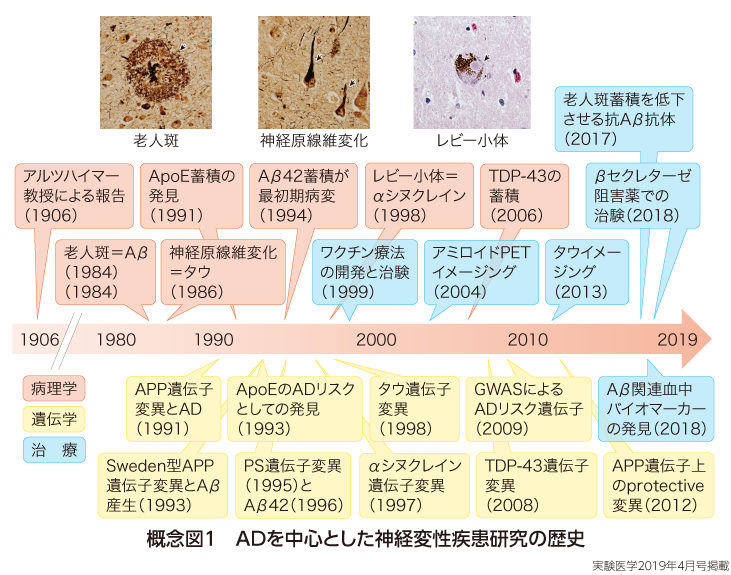
1980年代半ばから2000年代にかけ,これらの異常構造物と分子病態の理解が一気に進んだ.特にAD患者脳の生化学的な解析が行われ,まず老人斑の構成成分が,アミロイドβタンパク質(Aβ)であることが見出された.また患者脳から精製された画分に対する抗体を作出する手法を用いて,本邦の井原康夫らによりタウが神経原線維変化の主要構成成分であることが明らかとなった1).一方,1990年代から精力的に進められた家族性ADをはじめとする遺伝性疾患の解析により,Aβの前駆体タンパク質であるAPP(amyloid precursor protein)をコードする遺伝子上に点突然変異が見出され,Aβは生理的条件下でも脳内で産生されていること,さらにその遺伝子変異がAβ産生亢進や凝集促進をもたらすことが報告された.加えて,Aβのなかでも凝集性の高いAβ42が最初期病変であること,老人斑の構成成分として見出されたApoEが最も強い遺伝学的なAD発症リスクであることも明らかとなり,タンパク質の異常凝集と神経変性疾患の発症メカニズムが連関するものとして研究されることとなった2).この考え方を裏付けるように,家族性PDの原因遺伝子としてαシヌクレインが同定され,レビー小体の主要構成成分であることが示された3).またタウについても,その後家族性FTDの原因遺伝子として同定され,やはりその遺伝子変異がタウの凝集性を高めることが明らかとなった.そしてFTDの剖検脳において不溶化し凝集しているタンパク質としてTDP-43が同定され4),その後TDP-43遺伝子上にFTDに連鎖する点突然変異が見出された.すなわち,各疾患を病理学的に特徴づける異常構造物を構成するタンパク質の異常凝集と蓄積が,神経変性疾患発症の「セントラルパスウェイ」であるという概念の確立に至り(概念図1),Aβ,タウ,αシヌクレイン,TDP-43を分子標的とした創薬研究が精力的に続けられている5).
1AD治療戦略における抗Aβ療法開発研究から学んだこと
これらのなかで,最も研究が進んできたのは,ADにおけるAβを標的とした創薬ストラテジーである.特にAPPとは異なる遺伝子であるプレセニリン(PS)の変異でもAβ42産生を変化させること,PSがAβ産生酵素そのものの構成分子であることは病因分子としてのAβの位置付けを固めた.そして2012年に,ADや加齢に伴う認知機能低下を抑制するバリアントとして見出されたAPP遺伝子上のA673TバリアントがAβ産生を抑制することが報告され,脳内Aβ量を低下させることがAD治療薬となることの期待を一気に高めることになった6).
しかし前後して行われてきた,Aβ産生酵素であるβセクレターゼ,γセクレターゼ活性を抑制させる低分子化合物の治験は,大部分が失敗に終わっている.特にAβ産生量を規定する酵素であるβセクレターゼを阻害することでほぼその産生量をゼロとすることが可能であり治療効果が期待されていただけに,その失望感は大きなものであった.その原因についてはいまだ不明な点もあるものの,少なくとも治験に用いられたβセクレターゼ阻害薬は脳内に蓄積している老人斑の量をほとんど変化させることができなかったことが示唆されている.家族性AD遺伝子変異を利用した遺伝子改変マウスにおける前臨床試験では,Aβ産生を抑制することでアミロイド蓄積を低下させることが可能であり,マウスモデルとヒトでは大きく異なる.すなわち,ヒト脳においていったん蓄積してしまった老人斑は,何らかの理由でクリアランスが低下している可能性がある.
また老人斑蓄積のタイムコースが明らかとなり,ADが脳内における慢性的なAβ蓄積疾患であることが理解されるようになった.これは脳内における老人斑を可視化できるアミロイドPETの確立と,ADNIやDIANをはじめとする,年単位の大規模前向きコホート観察研究の成果によるところが大きい.そしてAD発症のはるか前から老人斑蓄積が認められること,また高齢健常者においても一定の割合で老人斑蓄積が認められ,そういった場合には数年後にADを発症するリスクが有意に高いことも明らかとなったのである.これらの結果から,Aβが蓄積して即座に神経細胞が変性するのではなく,5〜10年かけて神経細胞死に至っていくことが理解されるようになった7).すなわち,タンパク質蓄積病態が直接的に機能障害や細胞死を惹起する,という単純なスキームでは説明できないことが明らかとなった.したがって,脳内のアミロイド蓄積を早期に発見し,抗Aβ療法を開始する必要があると考えられており,そのために血液バイオマーカーの開発が期待されている(佐藤・岩田の稿)8).
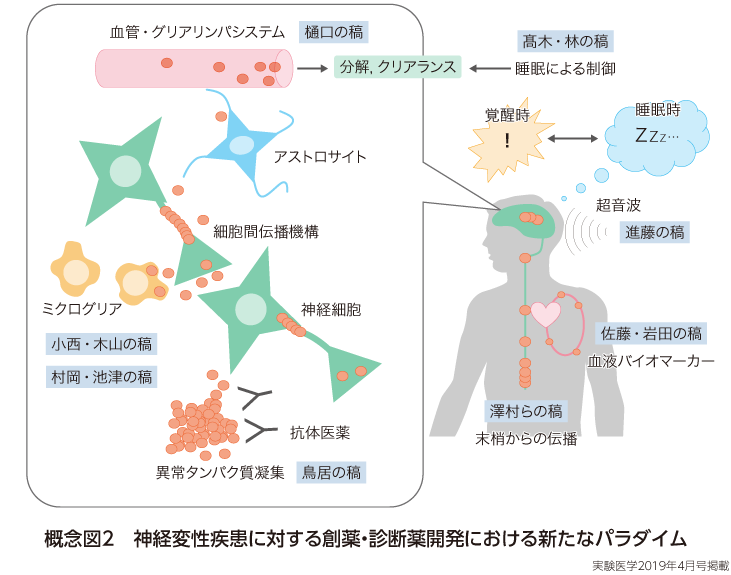
一方,新たな抗Aβ戦略として注目されているのが,抗体医薬である.そもそも20年前,高分子量タンパク質である抗体を用いて脳内の異常凝集タンパク質を除くという発想そのものは,非常識な考え方であった.しかし当時Elan社のSchenkらは,Aβそのものを能動ワクチンとして遺伝子改変マウスにインジェクションした結果,抗体価の上昇と脳内Aβ蓄積の抑制が相関することを見出したのである9).この結果は多くの議論を巻き起こしたが,その後,わずかではあるものの抗Aβ抗体が脳内へと移行し,おそらくミクログリアによる凝集Aβの除去,すなわちクリアランスを促すというメカニズムが示唆されている.また現在では受動ワクチン,すなわち抗Aβモノクローナル抗体による治療戦略が治験において検討されている.これは「常識」を打破する創薬基礎研究こそが,神経変性疾患克服に必要であることを示しているとも言えよう.近年では,新しい創薬モダリティとして注目されている核酸医薬や,非侵襲的なアプローチとしての超音波をADおよび神経変性疾患に適用する試みが始められつつある(鳥居の稿,進藤の稿,概念図2).
2疾患発症プロセスにおける新たなコンセプト
一方近年,この「セントラルパスウェイ」である異常タンパク質蓄積に連動して生じる新たな病態メカニズムが徐々に明らかとなりつつあり,その解明は革新的疾患修飾薬の開発につながることが期待されている.その1つに,遺伝学的解析の技術進歩がある.2010年代前後から,いわゆるゲノムワイド関連解析(GWAS)やエクソーム解析が爆発的に進展し,複数のADの遺伝学的リスク・予防因子が同定された10)11).しかし驚くべきことに,そのなかにはグリア細胞特異的な遺伝子や,Aβやタウの脳内動態(産生や分解)に直接関連しないものが数多く同定されたのである.すなわち,「神経」が変性するADにおいて,脳内のさまざまな「非神経」細胞が関連しながら病態を加速,もしくは遅延させることが示唆された12).これまで,AD研究の多くが神経細胞に着目し,非神経細胞における炎症性反応は,単なる異常タンパク質蓄積の結果と考えられてきた.そのため,これらの細胞がどのようなメカニズムによってAD発症プロセスに影響するかについては,現在の大きなホットトピックとなっている(概念図2).実際,非神経細胞であるアストロサイトやミクログリアにおける炎症反応や,血管系が関連する脳内における凝集タンパク質のクリアランス機構が大きな注目を浴びている(樋口の稿,小西・木山の稿).またこの脳内老廃物を排出するメカニズムが生理的には睡眠によって制御されていることが示され,睡眠の異常と神経変性疾患の発症に分子レベルでの相関が想定されている(髙木,林の稿).
加えて教科書的な知識を書き換える新たな生物学として研究されているのが,異常タンパク質蓄積病態の伝播である.もともと病理学的な解析から,ADにおいて細胞外に蓄積するAβと細胞内に蓄積するタウの,病態進行に伴う蓄積パターンが異なることが示されており,この2つの病理学的特徴を関連付ける機構が不明であった.また同様に細胞内に蓄積するαシヌクレインやTDP-43がなぜそれぞれ異なる領域に蓄積し,どのようにしてそれらのタンパク質がPDやDLB,FTDの各疾患において特徴的な神経変性を引き起こすのか,その分子メカニズムは不明であった.そういったなか,タウやαシヌクレインが神経回路に沿ってプリオン様に伝播することが示された.しかしこの現象は,「細胞質に蓄積」している異常タンパク質が,いったん「細胞外」へと放出され,さらに受容する細胞の表面膜を超えて再び「細胞質のタンパク質」の凝集を促す,という機序が想定されている13).この現象は古典的な膜輸送系を介したメカニズムでは説明ができず,新たなバイオロジーとして多くの研究者の興味を惹き,タウ伝播機構においてはミクログリアや細胞外小胞の関与が示唆されている(村岡・池津の稿).また中脳黒質の変性疾患であるPDでは,最初期のαシヌクレイン蓄積が嗅球や腸管における迷走神経から開始する可能性が報告されており,疾患概念そのものが変わりつつある(澤村らの稿).そしてこれら異常タンパク質の蓄積病態の伝播を阻害することで神経変性発症の抑制もしくは予防につながることが期待され,多くの創薬研究が進められている.
おわりに
神経の変性とともに異常構造物の出現という病理学的な特徴を示す神経変性疾患は,その生化学的な解析と遺伝学の解析が相まって,原因分子が明らかとなり,そこからバイオマーカー開発,そして大規模観察研究を経て,異常タンパク質の蓄積病態が長期間にわたって継続することで生じる,慢性代謝疾患であることが明確となった.ADにおいて抗Aβ医薬品の開発が難航している現状で,Aβが治療標的分子として正しくないのではないか,という議論も多くなされている.しかし遺伝学的な解析結果は明確にAβがADの発症にかかわることを示している.一方,タウの神経変性における重要性もまた,FTDの遺伝学解析から明白である.現代の生物学を研究するうえで,遺伝学を否定することはできない.つまり,ADにおいてはAβとタウ,どちらが正しいのではなく,ともに病態に深くかかわっている因子であり,合わせて研究していくことが求められているのである.
最近では,Aβのみならずタウも健常者において若年期から蓄積していることが明らかとなっている.このような蓄積と疾病発症の矛盾点については,脂質異常症におけるコレステロールおよび粥腫の病的役割を考えると,理解していただけるのではないだろうか.多少蓄積しただけでは,自覚症状は現れないが,長期間にわたって慢性的に病態が進行した結果,動脈硬化そして心筋梗塞などを招来する.改めて長期間にわたり,異常タンパク質が蓄積している状況をかんがみて,それぞれの実験モデル,創薬アプローチを見直す必要があるだろう.しかし一方で,これまでの失敗も含め,ADにおける創薬研究から得られた知見の多くは,PD,DLB,FTDなど,異常タンパク質蓄積を主因とする他の神経変性疾患においても適用可能であると考えられる.すなわち,未発症期もしくは超早期において疾患発症リスクを正しく見積もり,正しい創薬標的分子もしくは細胞にアプローチすることが,いずれの疾患についても必要である.
この30年間にわたる分子・細胞病態研究から,脳内の環境を整えるインフラストラクチャーとしての非神経系細胞の病態形成への寄与や,細胞間を伝播する細胞質内タンパク質凝集病態の伝播など,さまざまな新しいコンセプトが見出され,検証されつつある.また本特集では触れられなかったが,神経変性疾患における新しいバイオロジーとして,異常タンパク質凝集に関連した液-液相分離や,開始コドンを介さない翻訳(RAN translation)も注目されている.本特集を通じて,常に新しい挑戦を続けている神経変性疾患研究の醍醐味に興味をもっていただければ幸いである.
参考図書
- 「認知症 発症前治療のために解明すべき分子病態は何か?」(森 啓/編),実験医学増刊 Vol.35 No.12,羊土社,2017
文献
- Nukina N & Ihara Y:J Biochem, 99:1541-1544, 1986
- Selkoe DJ & Hardy J:EMBO Mol Med, 8:595-608, 2016
- Baba M, et al:Am J Pathol, 152:879-884, 1998
- Arai T, et al:Biochem Biophys Res Commun, 351:602-611, 2006
- Goedert M:Science, 349:1255555, 2015
- Jonsson T, et al:Nature, 488:96-99, 2012
- Sperling R, et al:Neuron, 84:608-622, 2014
- Nakamura A, et al:Nature, 554:249-254, 2018
- Schenk D, et al:Nature, 400:173-177, 1999
- Kanatsu K & Tomita T:Front Biosci (Landmark Ed), 22:180-192, 2017
- Takatori S, et al:Adv Exp Med Biol, 1118:83-116, 2019
- Heneka MT, et al:Lancet Neurol, 14:388-405, 2015
- Hasegawa M:Biomolecules, 6:doi:10.3390/biom6020024, 2016
著者プロフィール
富田泰輔:東京大学大学院薬学系研究科機能病態学教室教授,脳神経疾患治療学社会連携講座教授(兼任).1994年東京大学薬学部卒業,’97年より同大学院薬学系研究科助手,2000年同研究科学位取得,博士(薬学).’03年同研究科講師,’04〜’05年に米国ワシントン大学セントルイス校留学,’06年同研究科准教授を経て’14年より現職.専門は病態生化学.病気の基礎研究を通じて治療・予防・診断薬開発につながる成果を出すと同時に,新しい基礎生物学を切り拓きたい.