概論
空間トランスクリプトーム技術の最前線
Comparison of principles and specs in spatial transcriptomics technologies
沖 真弥,大川恭行
Shinya Oki 1)/Yasuyuki Ohkawa 2):Department of Drug Discovery Medicine, Kyoto University Graduate School of Medicine 1)/
Division of Transcriptomics, Medical Institute of Bioregulation, Kyushu
University 2)(京都大学大学院医学研究科創薬医学講座1)/九州大学生体防御医学研究所トランスクリプトミクス分野2))
多細胞システムは,空間的に整然と配置された多彩な組織や細胞型によって構成される.その細胞型の空間的な分布や固有の機能は遺伝子発現プロファイリングによって解析されるが,従来のin situ hybridization法や免疫染色ではすべての遺伝子を網羅するのは難しい.ごく最近,空間トランスクリプトームと総称される実験技術の開発により,網羅的な遺伝子発現プロファイルを空間情報と紐づけて解析できる時代が到来した.本特集では,さまざまな空間トランスクリプトーム技術の開発者に加えてユーザーにも寄稿してもらい,各技術の原理,スペック,導入コスト,長所や短所などについて紹介する.
はじめに
生物は,同一のゲノムをもつ多種多様な細胞により構成される.それぞれの細胞は,細胞型とよばれる“個性”を獲得し,組織において固有の機能を担う.この細胞型は,ゲノムからの選択的な遺伝子発現により形成される遺伝子発現の総体,すなわちトランスクリプトームにより決定される.したがって,トランスクリプトームの解析は基礎生物学研究の重要な切り口として汎用されてきた.近年の技術開発の加速により単一細胞レベルの解析が実現し,生体を構成する細胞型が網羅的に同定されつつある.一方で,特定の細胞型をもつ細胞は,疾病,加齢,運動あるいは栄養状態といった環境に応答して多様な細胞状態となる.これら,状態変化は,タンパク質,脂質,糖などの代謝産物の量,化学修飾や構造などの質,そして,空間的な局在変化として表れる.組織恒常性や機能破綻のメカニズムを明らかにするためには,従来行われてきた細胞型の決定に加えて,細胞状態の動態を明らかにする解析が求められている.
このような背景のもと,空間情報を保持したゲノム,エピゲノム,プロテオーム,メタボロームなどさまざまなオミクス解析の研究が進められているが,今回の特集では特に研究が進んでおり,汎用化されはじめている空間情報をもったトランスクリプトームに焦点を当てる.空間トランスクリプトームは,細胞の理解を,個々の細胞型に加えて,周辺の細胞の配置から理解する新たな試みといえる.当然,その解析には,単一細胞レベルの解像度で,網羅的な遺伝子発現を解析するより高度な技術が求められる.
本稿では,まだまだ発展途上ではあるがようやくその形がおぼろげながら見えてきた空間トランスクリプトーム技術について,開発の歴史を振り返り,現在の解析で可能になった点と開発途上な部分,そして今後の展開について触れてみたい.
1開発の歴史
2020年度,Nature Methods誌上でMethod of the Yearとして「Spatially-resolved transcriptomics」が選ばれた1).本法の画期的な点は,組織や細胞の形態といった画像情報とともに,位置情報に紐づけられたトランスクリプトーム情報を取得できる点にある.したがって,一見形態が同じ組織あるいは細胞であっても,トランスクリプトーム情報から細胞型や細胞状態を判別し,機能を再定義することが可能である.
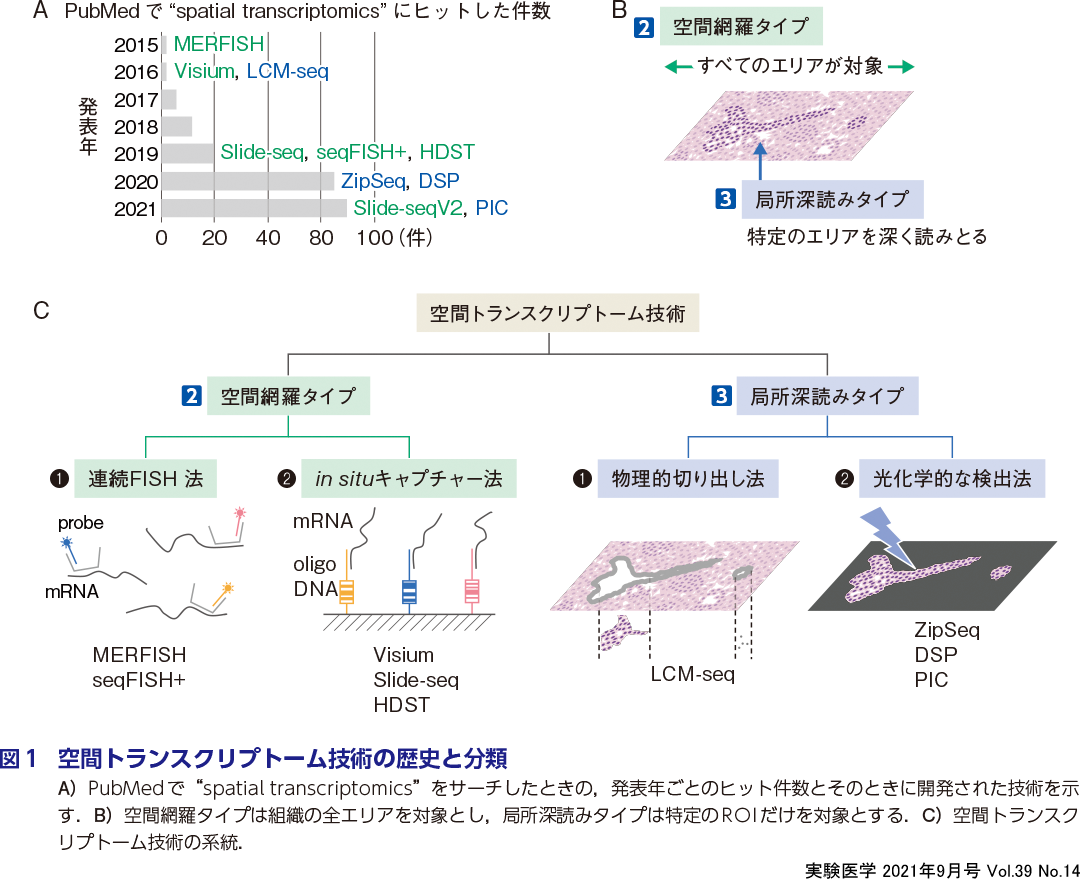
Spatial Transcriptomics という用語は2015年にはじめて論文に登場し,いま劇的な盛り上がりをみせている(図1A).Spatial Transcriptomicsと総称されるこれら技術は,Chenらが発表したfluorescent in situ hybridization(FISH)法を連続的に実施するMERFISH 2)を端緒とする.その後Ståhlらが,後述するin situキャプチャー技術による手法を用いてマウス脳切片全体の空間トランスクリプトームを取得する技術を「Spatial Transcriptomics」3)と発表したことで急速に広まり,しだいに一般名詞として定着していった.本技術は10x Genomics社によってVisiumという商標で商用化され,汎用化が図られている.これら技術開発の背景には,2000年代にマイクロアレイ技術,2010年代以降はRNA-seq技術によるトランスクリプトーム解析技術の発展がある.さらに分子バーコード技術の登場により,微量のRNAを定量的に増幅し,解析することが可能となった.また,単一細胞解析のデータ深度が劇的に高まり多くの機能未知の細胞型が相次いで同定され,組織中に含まれる細胞を新たに探索する機運が高まっていることもあげられる.
一方で,空間内の関心領域(region of interest,ROI)における遺伝子発現を網羅的に解析するための技術は以前から存在している.ROIに限定した発現情報を深く解析する局所深読みタイプの技術が発展した(図1B,C右).大きなものであればピンセットでROIを取り出せばよいが,サイズが小さくなれば熟練が必要となり,また取り出す前と後の画像情報を記録として残すことが難しい.このような物理的切り出し法はlaser capture microdissection(LCM)によって精度をはるかに向上でき,なおかつ画像情報を残すことができる.またごく最近,われわれやその他のグループから,光照射による化学的変化を利用した光化学的な検出法が開発され,より精細にROIを限定できる.一方で空間網羅タイプ(図1B,C左)はmmからcm単位に及ぶ広い空間を網羅するトランスクリプトーム技術である.これらは前述の連続FISHによるRNAイメージング法とin situキャプチャー法をベースとした技術の2つに分けられ,いま世界中で開発競争が激化している.
商用化されたものから,最先端の技術まで現在ではさまざまな技術が開発されているが,本稿では,空間トランスクリプトーム技術を2空間網羅タイプと3局所深読みタイプに大別し,それぞれのアプローチについて概念的な違いや課題点について論じる(図1).また,今後の展望について考えてみる.
2空間網羅タイプ
まずベースとなる技術から紹介する.in situ hybridization技術(ISH)は組織内の個々の部位(または細胞)でRNA分子を直接可視化することができる.これは,目的のmRNAに相補的な蛍光標識付きプローブをハイブリダイズ(以下,ハイブリ)させることで実現される.ISH自体は1960年代から存在していた技術であり,最も古典的な空間“トランスクリプト”技術である.一方で,より多くの異なる転写産物を同時に検出する場合,スペクトルの重なりが制限となってきた.これを解決する試みとして行われているのが,連続FISH法と,in situシークエンス(ISS)技術であり,ここでは ❶ RNAイメージング法と総称する.
連続FISH法にはMERFISH 2)やseqFISH+法4)があり,RNAを1分子レベルで蛍光観察するFISH(smFISH)をベースとし,数十回のハイブリで1万種類以上の遺伝子をイメージングできる.一方ISS技術は,in situでシークエンス反応を行うことでRNAを検出するアプローチである.連続FISHとISS法は,ハイブリダイゼーションとシークエンシングの違いはあるものの,RNAイメージングに基づくトランスクリプトームデータ取得という点で技術的基盤は比較的類似点が多い.
これに対して ❷ in situキャプチャー法は,バーコード配列を含むオリゴDNAの上に切片をのせてmRNAをキャプチャーし,RNA-seqでバーコードとcDNA配列を読みとる.トランスクリプトームデータの取得をイメージングから切り離すことでスループットを増やす方法であり,イメージングベースの方法とは一線を画している.いずれの空間網羅タイプのトランスクリプトーム技術の大きな魅力は,対象となる組織切片の全領域を対象とした遺伝子発現アトラスを得られることであろう.
❶ RNAイメージング法
1) 連続FISH法
MERFISH法は,たった16回のハイブリと消光のくり返しで1,000種類のmRNAを検出できる.またそののち登場したseqFISH+法では,80回のハイブリと脱プローブのくり返しで1万種類以上のmRNAの局在を解析できる(武井の稿).通常のsmFISH法では1,000種類のmRNAを検出するにはハイブリと脱プローブ作業を1,000ラウンドもくり返す必要があリ,マルチカラーにしたとしてもその回数が1/2から1/3になる程度である.少し難解ではあるが,ここではMERFISHやseqFISH+法がなぜ少ないハイブリ回数でこれほど多くのmRNAを検出できるのか,そのしくみについて簡単に説明したい(詳しくは武井の稿を参照).
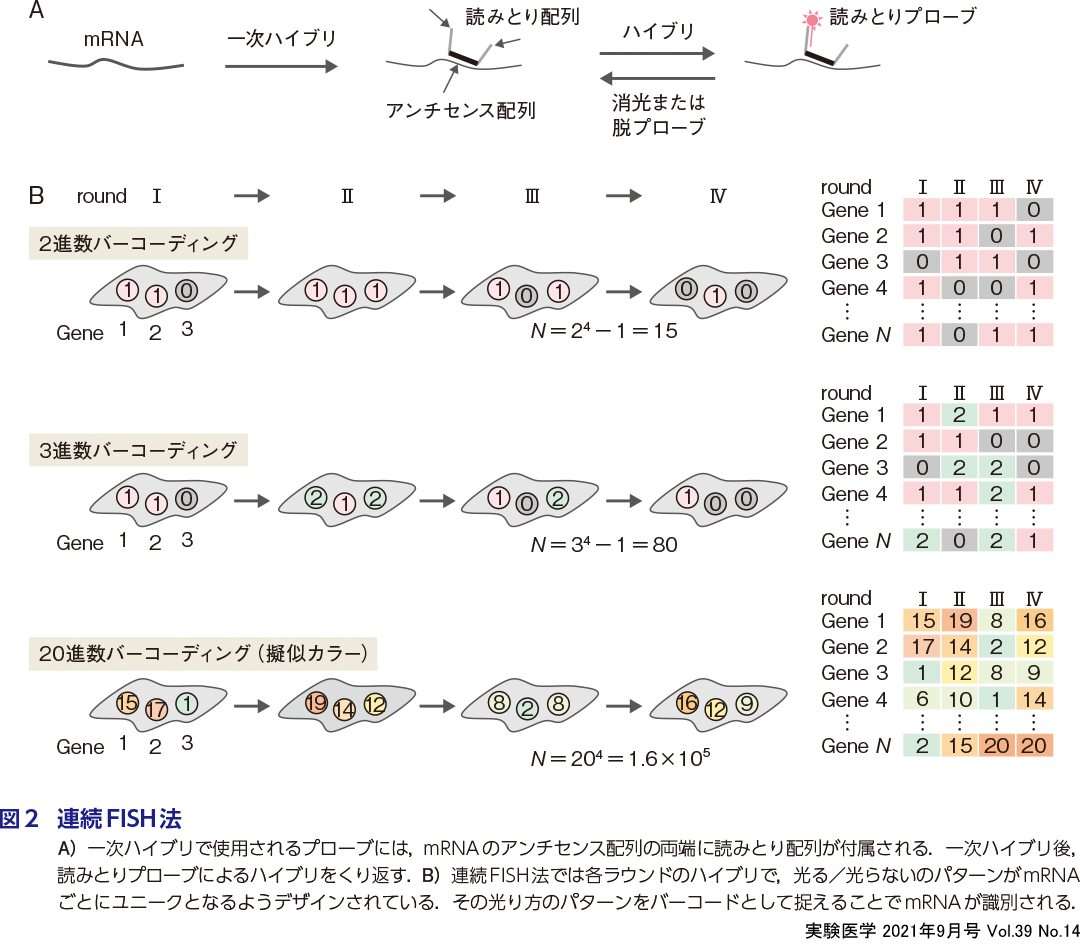
MERFISHやseqFISH+法ではまず,mRNAのアンチセンス鎖を含むオリゴDNA(一次プローブ)を使い,目的のmRNAにすべてハイブリさせる(図2A).この一次プローブには複数の「読みとり配列」が含まれ(MERFISHでは2種,seqFISH+では4種),mRNAごとにユニークな組合わせの読みとり配列が割り当てられるようにデザインされている.その後,蛍光ラベルした「読みとりプローブ」によるハイブリを何度もくり返し,蛍光顕微鏡で局在を逐次的に読みとる.シングルカラーで蛍光観察するMERFISHでは,各ハイブリラウンドで読みとりプローブがハイブリする(1)/しない(0),のどちらかを示すように読みとり配列がデザインされている(図2B上).つまり4ラウンド後にはmRNAごとにユニークな1または0の四桁の2進数が得られるため,最大で2 4-1=15種類のmRNAを識別できる(1を引くのは,全く光らないもの[0000]を除外するため).実際のMERFISHでは16ラウンド行うため,16桁の2進数が得られ,最大で216-1=65,535種類を識別できることになる.つぎに,2色の蛍光物質で読みとり配列を検出する場合を考えてみる.先と同様に,各ラウンドで読みとりプローブが赤(1)/緑(2)/ハイブリしない(0)のどれかになるように設計すれば,4ラウンドで四桁の3進数が得られるため,34-1=80種類のmRNAを識別できる.さらにseqFISH+法では四桁の20進数が得られるように設計しているため,20 4-1=1.6×105種類のmRNAを識別できる.しかしここで疑問が生じる.20進数を得るには20色もの蛍光プローブが必要だが,蛍光顕微鏡で20色を識別するのは不可能である.そこでseqFISH+法ではそれを「擬似カラー」というアイデアで実現している.これは1つのラウンドあたり20回のハイブリを行う.この20回のハイブリでは各mRNAが一度だけ光るように設計しているため,各mRNAに1から20の数字のどれかが擬似カラーとして割り当てられる.これを4ラウンドくり返すので,20 × 4 = 80 回のハイブリで四桁の20進数(20 4種類)が得られる.原理的には,1万種類の遺伝子であれば10進数の擬似カラー付けを4ラウンド(10 4)で十分だが,あえて擬似カラーを増やすことで読みとりエラーにも対応できるように工夫がなされている.
連続FISH法の課題点:
MERFISHやseqFISH+法は連続染色法をもとにし,原理的には全トランスクリプトーム解析をめざせる.しかし,いずれの手法も1分子のRNAを可視化して検出する場合,mRNAに複数のプローブをハイブリさせ輝度を上げる必要があり,原理的に短いmRNAを検出することは困難である.また,プローブ間の交差性を排除するためにはあらかじめ情報解析に裏付けされた精緻な設計が必要であり,さらにそのうえで,特殊なデバイスのセットアップが必要となるため安価な汎用的な技術となるまではまだ時間がかかりそうである.
2) in situシークエンス(ISS)法
ISS技術5)は,細胞内のRNAを直接シークエンス反応により検出する技術であり,原理的にはアンバイアスな空間トランスクリプトーム解析が可能である.まさに夢の技術といえる(原理の説明は難解であるため,本稿では割愛する).一方で,技術的には前述の連続FISH法よりも課題は多く,トランスクリプトームレベルの解析が可能かどうかは今後の開発しだいであろう.その理由は,まず配列決定に必要となるイメージングに十分な輝度を得るには,あらかじめ細胞内でRNAをDNAに変換し増幅させなければならない.細胞内の空間には限界があるため,現在の技術ではすべてのRNAを均等に増幅させることは実現していない.これら問題を回避する技術として,大きく分けて,技術的には後退するものの実用的な技術をめざしたバーコード読みとり技術としてのISS 5)〜7)と,困難ながらもアンバイアスな解析に挑戦するFISSEQ 8)のような流れがある.
❷ in situキャプチャー法
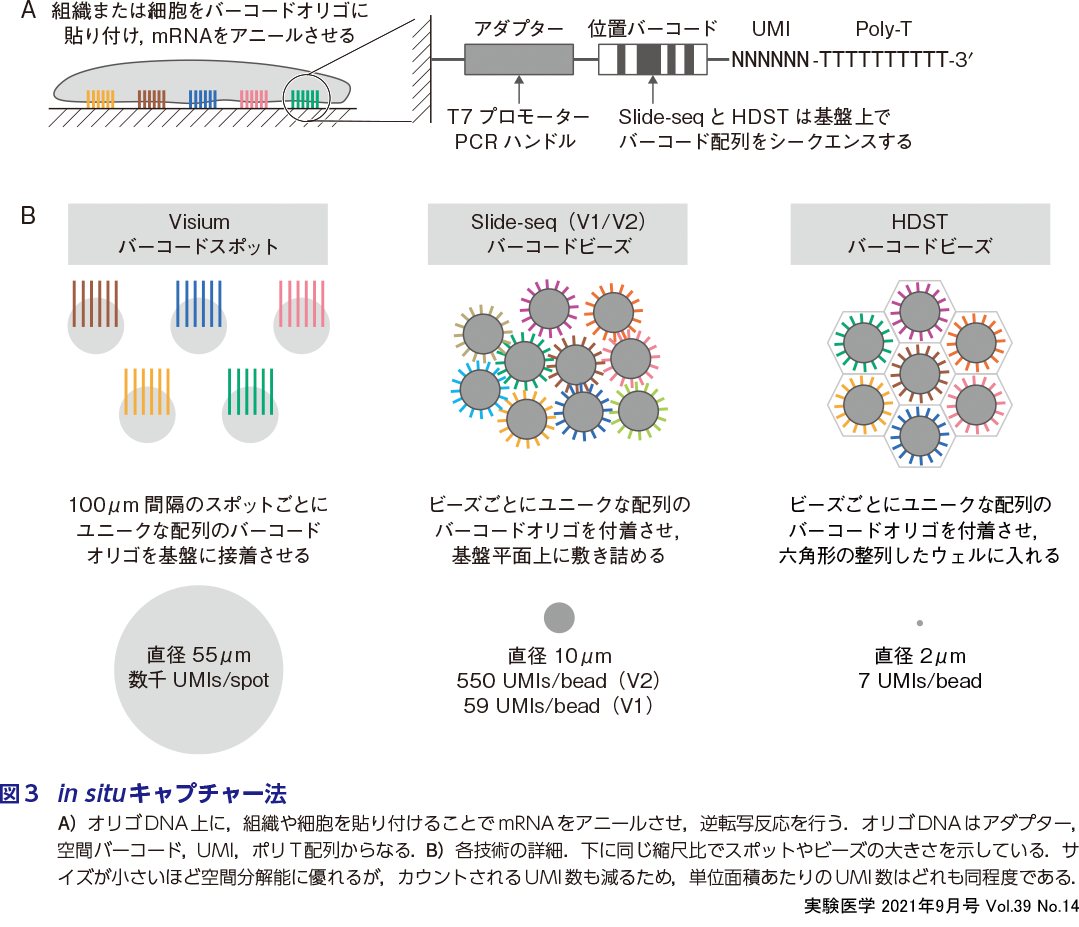
“魚拓方式”ともいえる技術である.組織切片に含まれるRNAをスライドガラス上の基盤に転写し,そのうえでライブラリー化することで基盤に設定された解像度のトランスクリプトームデータを得ることができる.最初にこの空間バーコード技術を用いた「Spatial Transcriptomics」法3)は,現在10x Genomics社よりVisiumというプラットフォームとして商用化されている.これは基盤上の直径55 μmのスポットにバーコード配列を含むオリゴDNAが植え付けられ,それが100 μm間隔で整列している(図3A,B).バーコード配列はスポットごとにユニークであり,あらかじめ座標ごとに決められている(水野らの稿).一方Slide-seq 9)10)やHDST法11)では,無数のオリゴDNAを付したマイクロビーズが基盤上にばらまかれる.ビーズごとにユニークな配列のバーコードが割り当てられており,その配列を基盤上でシークエンスしたのち,組織切片が貼り付けられる.つまり,バーコード配列と空間座標との対応があらかじめ決まっているのがVisium法で,あとからシークエンスで対応づけるのがSlide-seqとHDST法である.後者はそれぞれ10または2μmのビーズを使用することで,空間分解能を高めている.その後組織切片を貼り付け,組織像を写真として記録する.また,どの手法もオリゴDNAの末端にはpoly-T配列があるため,細胞内のpoly-A RNAとアニールし,細胞内逆転写反応が行われる.あとはcDNA:mRNAハイブリッドを回収し,VisiumとHDSTはin vitro transcription(IVT)で,Slide-seq法はテンプレートスイッチング法でcDNA増幅し,バーコード配列とともにシークエンスする.これにより,空間バーコードと遺伝子配列が対応づけられ,各遺伝子の発現量を座標ごとにマッピングできる.なお,すべてのオリゴDNAにはunique molecular identifier(UMI)という数塩基のランダム配列が付加されている(図3A).これにより,同じ配列のcDNAから同一のUMIが複数検出された場合,過剰増幅されたcDNAとして識別できる.つまりバーコードオリゴDNAにアニールした,増幅前のmRNAのコピー数をカウントできるため,正味の定量性が得られる.当初課題であった解像度も技術開発により著しく向上しており,今後最も注目する解析の流れであるといえる.
in situキャプチャー法の課題点:
魚拓方式ならではの課題として,組織サンプルからRNAを同一の効率で転写させることがきわめて難しい.まず,RNAの長さは分子種によって大きく異なり,高次構造も多様である.多くの場合,短いRNAほど転写されやすく長いRNAは転写されにくい.同一切片から取得されたRNA-seqと魚拓方式によるRNA-seqのデータに多くの場合相関性が乏しいことも課題とされている.これら課題はプラットフォームを提供する研究者(あるいは企業)側の課題というより,ユーザー側が手技の面で解決するべきポイントであり一定の負荷が求められる点であると考えられる.
3局所深読みタイプ
空間的な遺伝子発現情報を得る最もシンプルな方法は,単純にサンプル内から興味のある領域を分離することである.例えば,顕微鏡下で確認した組織領域をレーザー光で切り出す技術であるLCMは,最も普及している空間解析技術であろう12)13).また物理的な切り出しではなく,光照射によってROIに化学的な性質変化を引き起こす手法も開発されている.光活性化型プローブを生体に取り込ませるtranscriptome in vivo analysis(TIVA)14)やGFP等の蛍光タンパク質を発現させ細胞を分画させて解析するNICHE-seq 15)といったアプローチも開発され,1〜10個程度の細胞の解析に成功した.ただし,TIVAはプローブの取り込みが細胞膜の性状に影響されるという課題があり,NICHE-seqは遺伝的改変が可能なモデル生物への適用に限られるため,近年はより汎用性の高い光照射による手法が開発されている.ここでは特にLCMによる ❶ 物理的切り出し法と,光照射による化学的変化を利用した ❷ 光化学的な検出法に大別して概説する.
❶ 物理的切り出し法
1996年に開発されたLCM法12)は,細い径の高エネルギーレーザーで目的の細胞集団の輪郭を焼き切ることで,ROIを正確に切り出すことができる(図1C右).ROIの形状はコンピュータ上で自由に描け,そのとおりにレーザーが試料に射出される.切り出したROIの細胞からRNAを抽出すれば,トランスクリプトーム解析に供することができるため,2000年代初頭からマイクロアレイ,2010年代からはRNA-seqによる解析がなされてきた.またLCMの高精度化と,少数細胞RNA-seq技術の発展に伴い,2016年に発表されたLCM-seq法16)やGeo-seq法17)においては,シングルセルを切り出してSmart-seq2法による高感度RNA-seqに成功した.特にシングルセルから1万前後の遺伝子を検出でき,同一切片から複数のROIを切り分けて解析できる.
LCM法の課題点:
レーザーでROIを分離して解析する方法では,切り出せる領域の面積が微小になるほど難易度が増す.また切り出しには少なくとも数μm径のレーザーが必要なため,それ以下の空間分解能は得られない.レーザー出力が高ければ切り出しやすくなるが,照射痕が幅広くなるため,切り出しやすさと空間分解能のトレードオフについて留意されたい.
❷ 光化学的な検出法
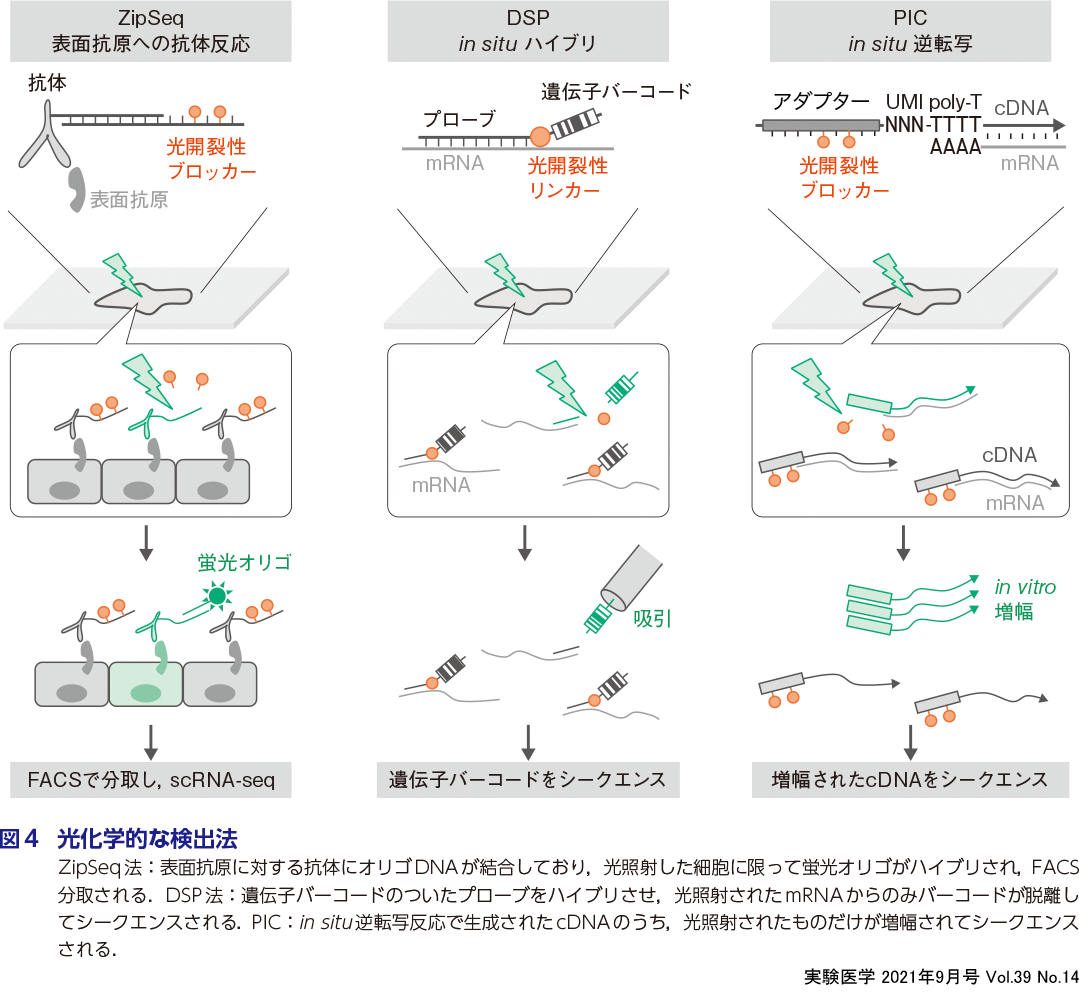
近年われわれやその他のグループは,光照射領域のみのトランスクリプトーム情報を引き出す技術を開発した.光は回折限界の数百nmの分解能で照射できるため,LCMよりも微細なROIにも対応できるのが特長である.ここで紹介する3つの技術(ZipSeq,DSP,PIC)は原理が異なるものの,光によって開裂するブロッカーやリンカー(正確にはケージド化合物とよばれる)を使用する点において共通している(図4).
ZipSeq法18)では,細胞表面抗原に対する抗体にオリゴDNAを結合したものを使用する.そこには光開裂性ブロッカーが結合しており,光照射で開裂した場合のみ蛍光オリゴDNAがアニールできるようにデザインされている.つまり組織切片に抗体を反応させたのち光照射すれば,ROIの細胞だけを蛍光ラベルできる.その後,組織を酵素でバラして,光る細胞だけをセルソーター(FACS)で分取しシングルセルRNA-seqを行うことにより,ROIに限定した発現プロファイルを1細胞レベルで取得できる.ZipSeq法は光照射のたびに異なるカラーの蛍光オリゴをアニールすることができるため,同一切片から数個のROIを分取できる.
NanoString社が開発したDigital Spatial Profiling(DSP)法19)では,mRNAのアンチセンスプローブに光開裂性リンカーとバーコード配列が結合したオリゴDNAを使用する.このバーコードは遺伝子ごとにユニークな配列としてデザインされている.これを組織にハイブリさせたのち光照射すると遺伝子バーコードが脱離するため,それを吸引してシークエンスすることでROIに限定した遺伝子発現プロファイルが得られる.DSPは専用の装置で光照射と吸引を何度でもくり返せるため,同一切片から数百ものROIを分離できる(植松の稿).
これらとは別に最近,われわれのグループが開発したPhoto-Isolation Chemistry(PIC)法20)では,光開裂性ブロッカーを結合した逆転写プライマーが使用される.これを組織に滴下して細胞内逆転写反応したのち光照射すると,ROIだけでブロッカーが脱離する.その後,組織ライセートをすべて回収しIVT反応を行うと,cDNA混合物のうちROI由来のものだけが増幅されてシークエンスできる.PICはその原理上,同一切片から複数のROIの分離はできないが,mRNAの検出感度が非常に高い(本田・沖の稿).
これら3つの方法では工程に免疫染色を入れられるため,ROIを厳密にラベリングできる.また光照射は通常の蛍光顕微鏡で行え,DAPIやHoechstの励起に使われる蛍光フィルターがあればよい.また任意の形状のROIに照射する場合は,市販されるデジタルミラーデバイスを励起光路に設置する.これは数百万個の微小ミラーの集合体であり,コンピュータの描画に応じてミラー角度が変化するため,描いたとおりのパターン照射が可能である.
局所深読みタイプの課題点:
DSPやPICは光を用いたプロファイリング法であり,簡便なアプローチで高解像度を実現している.深読みしつつスループットを上げることが理想的ではあるが,新たな概念的な転換が今後のハイスループットな手法開発へ向けた課題といえる.
4セレクションガイド
前述の技術のうち,どれを選ぶべきかについては,研究目的に沿って空間網羅性,深度,ROIの形状,コストなどを勘案していただきたい.ここではそのセレクションガイドをわれわれの考えで示すが,本特集では空間トランスクリプトーム技術の開発者やユーザーに寄稿していただいているため,各技術の詳細はそちらを参照されたい.
mm以上の広い範囲にわたる発現アトラスを作成したい場合は空間網羅タイプのうち,特にin situキャプチャー法がふさわしい.Visiumは商用化されているため,バーコードスライドを購入すれば簡便に実施できる(水野らの稿).Slide-seqやHDST法は商用化されていないため,バーコードビーズなどが必要となるが,Visium法よりも空間分解能に優れ,数個の細胞の違いを見極められる.ただし,どの手法もエリアあたりの検出深度は局所深読みタイプに比べて低い.スポットやビーズが大きければ検出できる遺伝子数も増加するが,どれも細胞あたりのUMI数は千個ほど,単位面積(μm2)あたりでは数個にとどまる.連続FISH法のなかでもseqFISH+法は検出深度に優れ,細胞あたり数万個,μm2あたり数十個のRNA分子を検出でき,細胞内局在まで理解できる.連続FISHのための装置とプローブが必要となるが,最新バージョンではDNAやタンパク質の分布も可視化できるのも特長である(武井の稿).またタンパク質の空間分布を解析したい場合は,数十種類であるが,Akoya Biosciences社のco-detection by indexing(CODEX)技術も検討されたい(宮脇の稿).
一方,ROIだけの発現情報を高感度で読みとりたい場合は,局所深読みタイプがよい.LCMによる物理的切り出し法は専用の顕微鏡と照射装置を必要とするが,100μm以上のサイズのROIであれば簡便に切り出せる.さらに小さく複雑な形状のROIには光化学的な検出法を検討されたい.空間分解能はZipSeqで1細胞レベル,DSPとPICは回折限界(数百nm)に達する.同一組織から複数のROIを解析するにはZipSeqやDSPがよい.ZipSeqは生細胞のスライスを,DSPとPICは組織切片を対象とする.DSPは商用化されており,光照射と吸引のための装置とプローブを購入すれば簡便に作業できる(植松の稿).PICはコストパフォーマンスがよく,使用するオリゴDNAは合成会社に委託(約10万円/数百回分)すれば誰でも入手でき,あとは既存法のRNA-seqを行えばよい.光化学的な検出法はどれも検出深度にも優れ,特にPICでは1細胞から70,000 UMI(~200 UMIs/μm2)が得られる(本田・沖の稿).
おわりに―現在の課題と今後の展開について
空間トランスクリプトミクス技術は非常に魅力的な組織解析技術になろう.一方で,ここで紹介した概念に基づく技術には共通の課題が存在している.その最も大きなものは,細胞の境界線の決定である.仮に,興味のある細胞があったとしても,その細胞が複雑な形態を示す場合や,組織内で入り組んだ構造に位置していた場合に,どの領域から切り出すか(あるいは光を照射するか),あるいはRNA分子を可視化あるいは転写した場合は,いったいどの細胞に帰属しているかを規定し,細胞単位の発現情報に転換するのは容易ではない.単純な画像解析では解決は困難であり,オミクス解析に匹敵する精緻な定量性を得ることはいまだ大きな課題といえる.
ただ,技術的には困難である一方で,空間トランスクリプトームは非常に魅力的な技術である.実際に解析することは多くのラボではかなりの困難が伴うであろうが,ぜひ取り組んでみてもらいたい.かつて次世代シークエンサーの導入の発展は,オープンソースの開発現場とそのコミュニティの発展に支えられた.本特集をきっかけに多くのユーザーが生まれ,分野全体が発展することでその先にある,単一細胞レベルでの全細胞レベルの生体機能の理解につながることを期待したい.
文献
- Marx V:Nat Methods, 18:9-14, 2021
- Chen KH, et al:Science, 348:aaa6090, 2015
- Ståhl PL, et al:Science, 353:78-82, 2016
- Eng CL, et al:Nature, 568:235-239, 2019
- Ke R, et al:Nat Methods, 10:857-860, 2013
- Chen X, et al:Nucleic Acids Res, 46:e22, 2018
- Wang X, et al:Science, 361:doi:10.1126/science.aat5691, 2018
- Lee JH, et al:Science, 343:1360-1363, 2014
- Rodriques SG, et al:Science, 363:1463-1467, 2019
- Stickels RR, et al:Nat Biotechnol, 39:313-319, 2021
- Vickovic S, et al:Nat Methods, 16:987-990, 2019
- Emmert-Buck MR, et al:Science, 274:998-1001, 1996
- Simone NL, et al:Trends Genet, 14:272-276, 1998
- Lovatt D, et al:Nat Methods, 11:190-196, 2014
- Medaglia C, et al:Science, 358:1622-1626, 2017
- Nichterwitz S, et al:Nat Commun, 7:12139, 2016
- Chen J, et al:Nat Protoc, 12:566-580, 2017
- Hu KH, et al:Nat Methods, 17:833-843, 2020
- Merritt CR, et al:Nat Biotechnol, 38:586-599, 2020
- Honda M, et al:Nat Commun, 12:4416, 2021
著者プロフィール
沖 真弥:2000年,大阪大学工学部卒業.’07年,大阪大学大学院医学系研究科 博士課程修了.博士(医学).同年より九州大学大学院医学研究院で助教,講師を経て,’20年4月より現在に至る.JSTさきがけ研究員(兼任).大学院生のときから10年以上にわたってマウスの発生生物学の研究に没頭し,100μmほどのマウス胚の胚葉を切り分ける神技をもつ.日々,腕とピンセットを磨いてきたが,神技に頼るのは科学の根幹である「他者による再現性」が担保されないと考え,自らそれを捨て,誰でもできるPICのアイデアに至った.時空間的な遺伝子発現の不思議に迫り,発生現象の神秘を解き明かすことを目標としつつ,疾患の理解などにも応用したい.
大川恭行:1997年,岡山大学工学部卒業.2003年,大阪大学大学院医学系研究科 博士課程修了.博士(医学).’03年より米国マサチューセッツ大学にてポスドク.クロマチン研究をはじめる.’06年より九州大学高等研究院にテニュアトラック准教授として独立,’11年テニュア取得,’16年に現職,生体防御医学研究所トランスクリプトミクス分野教授として着任.主に骨格筋の再生,老化のメカニズムをクロマチン構造レベルで理解することを目的として研究をしている.研究に必要な技術があれば,どのような技術でも開発することをモットーとしている.現,日本筋学会理事.