- [SHARE]
- ツイート
")

はじめに
ゲノム編集とは,生物のゲノムDNA上の狙った遺伝子領域を切断して,人工的に特定の遺伝子に対して置換,削除,挿入などの操作をすることであり,分子生物学者にとっては,ゲノム上の遺伝情報を自在に書き換える夢の遺伝子操作技術であるといえる.ある遺伝子の機能を知りたいときには,その遺伝子が破壊された変異体を野生型生物と比較して,どのように表現型が変化するかを調べる遺伝学的手法が有効である.生物が有する相同的DNA組換え能を利用して標的の遺伝子を改変するジーンターゲティング技術は,それが比較的容易な微生物の一部のモデル生物ではさかんに用いられてきた.さらに高等真核生物にも利用され,1989年にノックアウトマウス作製技術が開発されて (2007年ノーベル生理学・医学賞),作製までの煩雑な操作と長時間を要するにもかかわらず,その有用性により多くの研究に利用されてきた.ゲノム上の特定部位を狙って二本鎖切断を起こすことができれば,ノックアウトマウス作製で開発された技術に比べてはるかに簡単にゲノム編集ができる.制限酵素のFokⅠのヌクレアーゼドメインを利用して,人工的にZnフィンガーモチーフを融合させたZFN (zinc-finger nuclease) や,キサントモナスの転写エフェクターであるTALE (transcription activator-like effector) タンパク質のDNA認識モジュールを結合させたTALEN (TALE nuclease) といった人工ヌクレアーゼを利用したゲノム編集操作は,CRISPR/Casシステムの登場により大幅に簡略化され,利用範囲が広がって一気に開花した.ZFN, TALENおよびCRISPR-Casを利用したゲノム編集操作の原理については,最近の総説1)2)に詳細に解説されているので,ここでは触れない.
CRISPR発見のきっかけ

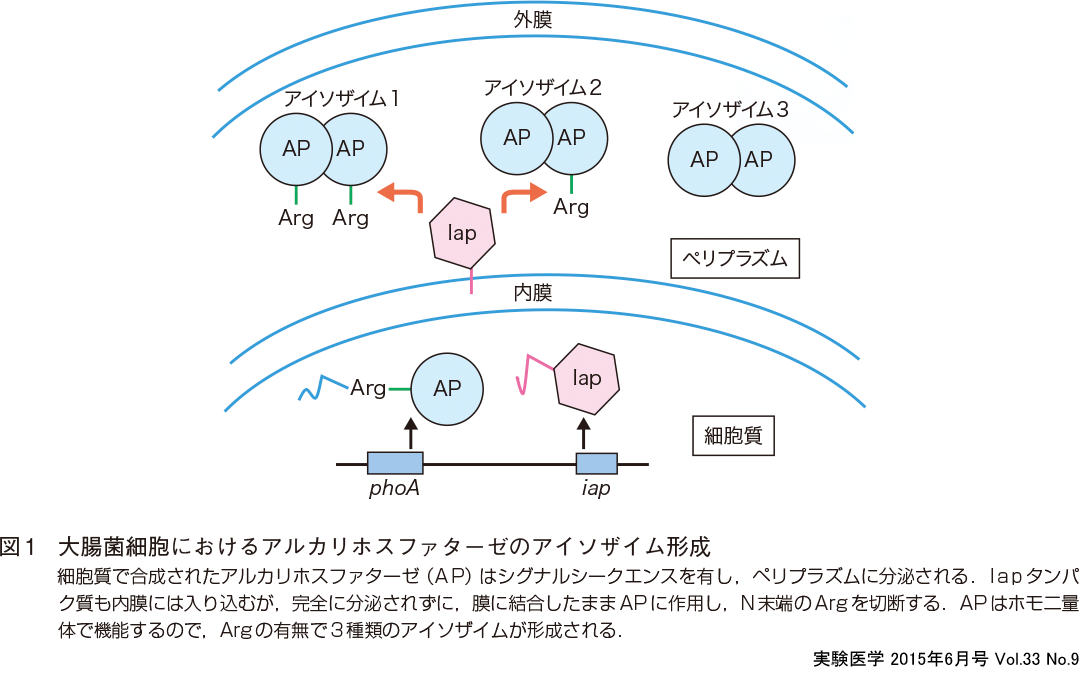
筆者は1983年から大阪大学微生物病研究所の化学療法部門 (当時) に所属し,大腸菌のDNAリガーゼ遺伝子 (lig) を同定し,その多量発現系を構築して,1986年に博士論文にまとめた後,当時の指導教官であった中田篤男先生がライフワークにされていた大腸菌のアルカリホスファターゼに関する研究に加わった (写真1).アルカリホスファターゼ (AP) はホモ二量体で機能する酵素であるが,細胞内においてサブユニットタンパク質のN末端のアルギニン (Arg) の有無により3種類のアイソザイムが存在する (図1).アイソザイム間での酵素活性は変わらない.このアイソザイム形成の生理的意味は不明であったが,アイソザイム形成が起こらない変異株が単離され,その原因遺伝子が存在することがわかりiap(isozyme conversion of alkali phosphatase) と名付けられた3).筆者はAPのアイソザイム形成機構を解明すべく,まずiapの塩基配列解読にとりかかった.これがCRISPRとの出会いにつながる.
DNAシークエンシング,その初期のころ
当時のDNAシークエンシングは,配列を読みたいDNAをM13ファージDNAで構築されたベクターを用いてクローニングし,そこから一本鎖DNA調製を行って鋳型DNAを用意した後,市販されて急速に普及していたシークエンスキットを用いて,ジデオキシチェーンターミネーション反応を行うという操作であった.もちろん微量のDNAは見えないので,放射性同位元素32PでDNAを標識する必要があり,そのためシークエンシング操作は放射性同位体使用可能な管理区域内で行う必要があった.一つのDNA試料につき,A,G,C,Tの4種のジデオキシ反応を独立に行った後,8 Mの尿素を含んだポリアクリルアミドゲル電気泳動で一ヌクレオチドの長さの違いを分離する.この電気泳動のゲルを作成するガラス板は20 cm × 50 cmが標準で,このゲルに一試料につき4種のジデオキシ反応液を泳動するので,20 cmの幅では,一度に6試料しか泳動できなかった.2時間以上かけて電気泳動した後,ガラス板を剥がし,ポリアクリルアミドゲルを乾燥させて,X線フィルムに接触させてオートラジオグラムをとることによって,漸く電気泳動結果が画像として読みとれるようになる.しかし,この操作で確実に解読できるのは150塩基程度である.解読範囲をできるだけ伸ばしたいために,もう1枚20 cm × 65 cmのゲルを用意して,2枚のゲルに同じ反応液を泳動して,2.5時間で短い方の電気泳動を終了し,もう1枚はさらに4時間泳動することにより,より長い領域を解読すると,合わせて350〜400塩基程度が読みとれた.ジデオキシチェーンターミネーション反応はプライマーの3′末端から開始されるので,長鎖のDNA配列を解読するためには,鋳型DNAを変えずに解読とプライマー合成をくり返すプライマーウォーキング法が単純に考えやすいが,今と違ってプライマーに用いるオリゴDNAを化学合成するのは高価であり,また,配列情報がないとプライマー合成ができないので,同時にいくつものサンプルを解読することができない.そのため,ベクターDNA中の特定の箇所に張り付くユニバーサルプライマーを利用し,その下流に解読したいDNAをクローニングした後に,解読したいDNA断片をプライマーに隣接した端から段階的に150ヌクレオチドごとに一定方向に削っていく操作をした.このようにして1〜2 kbpの長さの遺伝子でも全長配列を解読することができた.
当時の日課としては,朝から大きなガラス板を4枚用意してプレ泳動をはじめる,12試料にジデオキシ反応を仕掛ける (12×4=48反応),そして,電気泳動が終わったらゲル乾燥機にかけて,乾いたゲルをX線フィルムと重ね合わせて感光させる.この操作を1日12時間以上かけて行い,感光したフィルムを翌朝現像してオートラジオグラムができあがる.暗室のなかでフィルムを現像液,定着液に浸して行き,安全光のなかで,塩基配列が読めていることを示すバンドのラダーが見えたときに快感を憶えた経験は,筆者と同世代の分子生物学者は皆理解いただけるのではないだろうか.ガラス板のゲル電気泳動を使わない蛍光式キャピラリー法以降に分子生物学をはじめた若手の方々には,この一連の操作のたいへんさを想像できないと思うが,この実験には,DNAをとり扱う分子生物学実験操作がぎっしり詰め込まれている.この操作をきっちりこなせるようになれば,どんな分子生物学実験でも適切に操作できると思われる.筆者は近年,研究室に入ってきて分子生物学実験をはじめる初心者の学生にとって,この操作によってきれいなシークエンスラダーの画像がとれるようなるまでしっかりトレーニングを積めば,その後どの領域の研究に進んでも適切に対処できると考え,当研究室の学生には毎年くり返し話している.
[オススメ]医学・生命科学・農林水産学で必須の実験技術.様々なツール・生物種を網羅した決定版
iap遺伝子の配列解析
さて,話をiap遺伝子に戻そう.筆者は前述のような操作によって,DNAリガーゼをコードするlig遺伝子の全配列 (671アミノ鎖からなるタンパク質コーディング領域を含む2,552塩基長) を解読した実績をもとに4),iap遺伝子の配列解読に臨んだ.上記の操作によって,結果的に1,664塩基を解読したことになる.当時,DNAの配列を解読して論文として発表する際に,解析データの基準としてDNA二本鎖の双方を独立に端から端まで解読することが要求されていた (片方の鎖を読んで,相手側を相補的に推定するのではない!).筆者も当然その基準をクリアーすべく実験をくり返した.間違った配列を発表しないためにもこの基準はきわめて重要であったと思うが,実際にはなかなか思うように進まないのが実験である.当時のジデオキシ反応には大腸菌のDNAポリメラーゼⅠからN-末端の5′-3′エキソヌクレアーゼドメインを欠失させたKlenow酵素が用いられていた.今のような耐熱性のポリメラーゼではないので,37℃でDNA鎖合成反応を行うと,鋳型DNAが自己的に二次構造などを形成しやすく,反応の進行を阻害する.そのためにジデオキシヌクレオチドのとり込みによらない反応停止が起きると,その部分の配列は確定できない.その場合はM13ベクターにクローニングするDNA断片の切断位置を変えて,プライマー末端に続く読みはじめの位置を変えながら解読できるまで実験をくり返す必要があった.得られたオートラジオグラムのバンドを1本ずつ目で読みながら,手動でA,G,C,Tをコンピューターに打ち込んでいく.独立に両鎖の配列を解読し,最後に両鎖の相補性が完全に一致すれば,晴れて解読終了となる.
iap遺伝子の解読のときは,現像したX線フィルムをシャーカステンの上に置いて,私がバンドを読み上げて,それを隣で中田先生がコンピューターに打ち込んでくださった.一塩基も読み間違えずに,1日も早く解読したいという先生の意気込みを肌で感じていた.二人並んで机の前に座り,熱中した光景を今でも鮮明に思い出す.このようにしてiap遺伝子の塩基配列を読み終え,翻訳の読み枠を確定し,iap遺伝子産物が345アミノ鎖からなるタンパク質であることが判明した.このタンパク質が大腸菌の内膜に結合した状態で,ペリプラズム内に分泌されるアルカリホスファターゼに作用して,N末端のArgを切断するという結果と合わせて論文を執筆した5)(図1).
CRISPR発見
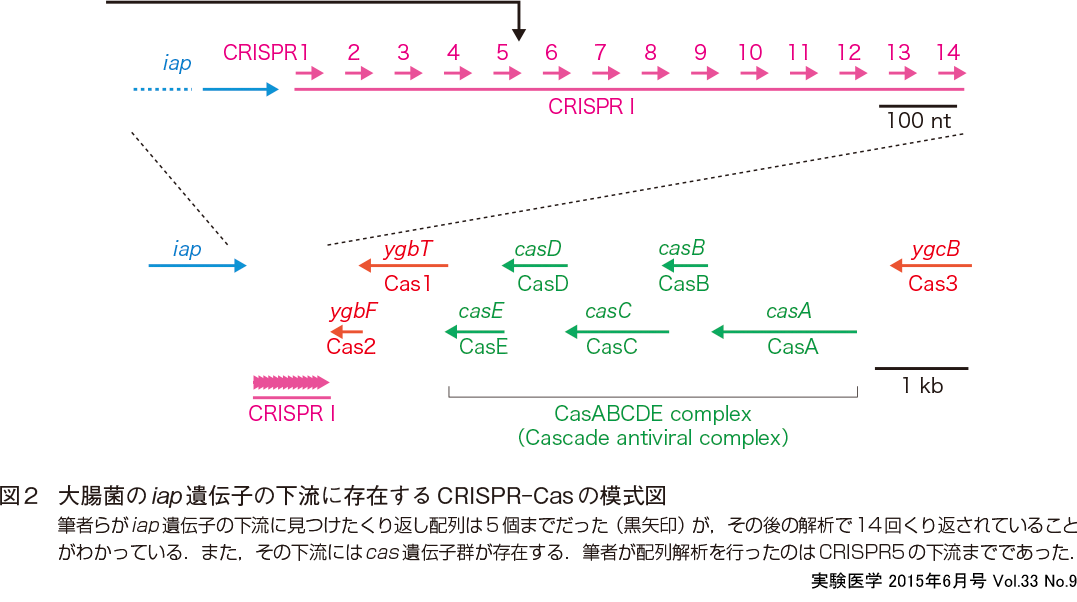
前述のようにしてiap遺伝子配列を解読したが,ベクターに組込まれていたDNA断片にはiap遺伝子の翻訳終了の終止コドン (TGA) の下流にまだ300塩基ほど含まれていた.この領域は,高次構造をとりやすい配列だったようでKlenow酵素を用いたジデオキシ法で決定するのはたいへんな骨折り作業であった.解読可能なオートラジオグラムがなかなか得られず,鋳型DNAを何度も作製しなおして実験をくり返した.iap遺伝子の翻訳領域は読み終わっていたので,下流の部分はそこまで正確に解読しなくてもいいのではないかと思ったこともあったが,クローニングされたDNA断片を完全に読みあげたいという思いで粘り強く実験した.両方の鎖を読み終えて,中田先生と一緒に2本の配列を重ね合わせたときに,A-T, G-Cの相補性が完全にマッチしたときの達成感も忘れられない.この領域が特徴的で,転写終了のためのターミネーターと想像されるステムループ構造形成可能なパリンドロミック配列の下流に規則正しいくり返し配列が存在した (図2).この配列は29ヌクレオチドからなり,32ヌクレオチド長のスペースを挟んで5回くり返していた.くり返しのコンセンサス配列は5′-CGGTTTATCCCCGCTGGCGCGGGGAACTC-3′というもので,2回対称性の14ヌクレオチド配列が含まれる.顕著に特徴的な配列であったが,これが生理的に何を意味しているのかわからず,当時知られていた,バクテリアに保存された二回対象のREP配列がmRNAの安定性に寄与することを上げて議論するのが精一杯であった3).
このくり返し配列の意味がわからないまま,筆者はポスドクとして1987年8月にYale大学へ赴任した.頭のなかはすっかりアメリカでの研究生活のことで一杯になっていた.はじめて経験する海外での生活のセットアップと,そこでとり組むtRNA依存Glu-Gln変換機構の研究テーマのことで考えることが多く,全く余裕のないときに,iap遺伝子の論文がJournal of Bacteriologyに受理された知らせをいただいた.これが今,頻繁に引用していただいているCRISPR発見の論文である3).論文が受理されたときは,はっきりと気持ちが元気になり,頑張って新しい研究テーマを進めようと活気づいたことを思い出す.しかしこの論文のDiscussionで記載されている奇妙なくり返し配列のことは,そのときも,またその後も忘れてしまっていた.中田先生はその後,この配列が大腸菌のゲノムのなかでさらに続き,合計14回くり返されること (図2),またこのようなくり返し配列は大腸菌のゲノム中にもう一カ所存在すること,さらに,これがサルモネラや赤痢菌にも存在することを確かめられて発表された6).
CRISPRの機能解明から応用へ
1990年代後半からゲノム解析の時代に入り,比較的ゲノムサイズの小さいバクテリア,アーキアは,相次いで全ゲノム配列が解読されてデータベース上に登録されるようになった.その結果,原核生物のゲノム上には,このような特徴的なくり返し配列が広く存在することがわかってきた.この配列を統一的にCRISPRとよぼうという提唱が2002年になされ,さらにCRISPRの近傍には保存された遺伝子クラスターが存在することがわかり,機能的にCRISPRと連動していると予想してCas (CRISPR-associated) geneと名付けられた7).その後,バイオインフォマティクスによる詳細な配列比較の結果,CRISPR-Casの生理的機能としてファージ感染からの防御やプラスミドによる形質転換の抑制を担う原核生物の獲得免疫機構が提唱された8).この仮説は2007年に乳酸菌の一種であるStreptococcus thermophilusを用いて実験的に証明され9),原核生物の獲得免疫システムとして広く知られるようになった10)11).そして,CRISPR-Casによる部位特異的なDNA切断機能をゲノム編集に利用した報告が2013年に出て12)13),ゲノム編集操作が一気に開花し,現在急速に広がっている.
おわりに

私が約30年前に大腸菌の遺伝子解析から発見した独特のくり返し塩基配列は,CRISPRと名付けられて,ゲノム編集技術に応用されるに至った.奇妙なくり返し配列の発見の論文は,最近のCRISPRやゲノム編集を紹介した論文で頻繁に引用されている.また,そのために旧友からしばしばメールや電話をもらうようになったことが増えた.筆者にとってはたいへん嬉しいことである.それだけゲノム編集の注目度が高く,CRISPRのゲノム編集技術への貢献のインパクトが大きいということである.筆者はこのくり返し配列の研究を続けなかったが,配列解析操作を行いながら,このくり返しには何の存在意義があるだろうと思っていたし,論文のなかでも特筆してその生理的意味はわからないと記述している.筆者はアメリカでのポスドクを終えて帰国した後は,超好熱性アーキアのDNA複製,修復の研究をはじめた.この領域も当時ほとんど未開の地であったので,切り開いていくなかで何度も興奮を味わうことができた.アーキア研究領域では世界中に友人ができ,ゴードンカンファレンスや極限環境微生物の国際会議などで頻繁に交流を続けている.アーキアの研究領域でも数年前からCRISPRの研究が目立ち,今では独立したセッションが組まれるようになった.研究者の間で,いつも全く異なる内容を発表している私がCRISPRの発見者であったことの偶然をおもしろがって,Mr. CRISPRとよんでくれる友人もいる (写真2).
今あらためて強く思うことは,CRISPR発見当時は配列情報しかなかったが,それが何か特徴的であるということは,偶然ではなく意味をもつということである.CRISPRに限らず,生命科学においてそれまで知られていることと違う何か特徴的なものを見つけたときには,大発見,大発明につながるので,環境が許されるならかかわりをもって根気よく研究を続けることが重要だと思う.大学には,すでに高評価されている対象や,役に立つ出口が見えているテーマの研究ばかりではなく,意味のわからないくり返し配列のような研究でも,地に足をつけてじっくりと続けられるような環境がきわめて大切なのだ.
文献
- 1) Jinek M, et al:A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science, 337:816-821, 2012
- 2)「CRISPR/Casが生命科学を加速する」(畑田出穂/企画),実験医学Vol.32 No.11, 2014
- 3)Nakata A, et al:Gene, 19:313-319, 1982
- 4)Ishino Y, et al:Mol Gen Genet, 204:1-7, 1986
- 5)Ishino Y, et al:J Bacteriol, 169:5429-5433, 1987
- 6)Nakata A, et al:J Bacteriol, 171:3553-3556, 1989
- 7)Jansen R, et al:Mol Microbiol, 43:1565-1575, 2002
- 8)Makarova KS, et al:Biol Direct, 1:7, 2006
- 9)Barrangou R, et al:Science, 315:1709-1712, 2007
- 10)Horvath P & Barrangou R: Science, 327, 167-170, 2010
- 11)Wiedenheft B, et al:Nature, 482:331-338, 2012
- 12)Cong L, et al:Science, 339:819-823, 2013
- 13)Mali P, et al:Science, 339:823-826, 2013
著者プロフィール
- 石野良純
- 1981年,大阪大学薬学部卒業,’83年,大阪大学大学院薬学研究科前期課程修了/同年宝酒造入社,大阪大学微生物病研究所研究生 ’86年,薬学博士,’87年,Yale Universityポスドク,’89年,宝酒造バイオプロダクツ開発センター,バイオ研究所主任研究員,’96年,(国プロ) 生物分子工学研究所主任研究員,主席研究員を経て2002年より現職.研究テーマ:修士論文は制限酵素に関する研究,博士論文はDNAリガーゼに関する研究,ポスドクはtRNA依存アミドトランスフェラーゼに関する研究,帰国後は現在までアーキアのDNA複製と組換え修復機構,およびDNA関連酵素の遺伝子工学技術開発への応用に従事している.資格:薬剤師,第一種衛生管理者.