概論
クライオ電顕が構造解析に革命を起こしている
Cryo-electron microscopy as a new tool for structure biology and drug design
佐藤主税
Chikara Sato:Biomedical Research Institute, Structure Physiology Research Group, National Institute of Advanced Industrial Science and Technology(AIST)(産業技術総合研究所バイオメディカル研究部門構造生理研究グループ)
構造生物学は今激動の時代にある.溶液中に分散したタンパク質を凍らせるだけで観察できるクライオ電子顕微鏡(電顕)が進化し,単粒子構造解析法が原子分解能に到達した.この方法は試料の結晶化を必要としないため,難結晶性なタンパク質でも,精製品が少量あるだけで構造決定への道が拓かれることを意味する.現状では,はっきり写る大きなタンパク質を得意とする方法であるが,解析可能な分子量も下がってきている.例えば,比較的小さな分子であるGPCRなどでも解析例が出はじめており,将来創薬ターゲットタンパク質の多くが解析可能な領域に入ると期待される.本クライオ電顕特集では,単粒子解析法と(電子線)トモグラフィー法の原理とその現状を解析例と共に解説し,システム利用のコツ・専門家なら知っている常識・システム導入の注意点も説明したい.
はじめに
生命の機能を成り立たせる根幹は生体分子とその複合体である.それらの構造は,解明されるだけで分子機構を直感的に示唆し,細胞メカニズムの理解を大幅に進めることがある.DNAの2重らせんの構造決定が,その顕著な例であろう.もちろん,すべての構造解明が機構の理解へと直結するわけではない.しかし,新たな構造決定に挑戦する研究者には,そのようなロマンを抱く人も多いと思う.近年の構造生物学は,三次元結晶を用いたX線結晶解析,NMR法などの進歩により大きな成果を得た.しかし,結晶を作製しにくいタンパク質も多く,結晶化のプロセスを必要としない解析法が求められていた.そのようななかクライオ電子顕微鏡による単粒子解析法が原子分解能に到達したことで,構造生物学は大きな変革期を迎えつつある.単粒子解析法では,結晶ではなく,溶液中に散在するタンパク質粒子を薄い層中に瞬間凍結することで,さまざまな方向を向いた粒子を電顕撮影する.得られた画像より数万個程度の粒子像を拾い上げ,そこから三次元構造を決定(再構成)する.そのため,精製量は,X線結晶解析に比べ4ケタ以上少なくてよい.本法により,これまで結晶化が難しかった膜タンパク質も構造が続々と解けはじめた.また,構造変化がダイナミックすぎるがゆえに結晶を作製しにくいさまざまなタンパク質群もターゲットに加わった.なかでも,創薬ターゲットの約半数を占めるGタンパク質共役型受容体(GPCR)は,動的で容易に多状態をとりやすく,単一構造による結晶は容易には形成されない.そのため,単粒子解析法による構造解析への期待は大きく,今後ますます普及することが予想される.
1本特集の全体像(概念図)

本特集の構成について説明する.概論では単粒子解析法の原理と作業のおおよその流れを概説する.また,この分野を熟知する専門家なら知っている単粒子解析法のコツと要点,システム導入の注意点・難点も説明したい.まずはじめの3稿ではクライオ電顕にまつわる3つの技術革新を紹介したい.光岡・横山の稿では実際の解析成功に最も決定的な役割を果たすサンプル調製法の進歩を,岩崎の稿では単粒子解析法が原子分解能に到達する原動力となったハードウェアの進歩を,安永・荒牧の稿ではソフトウェアの進歩の道のりを,原理と応用とともに一線のクライオ電顕学者達が解説している.これら新たな生化学手法・装置・ソフトは,さらなる高分解能へ向けて進歩を続け,新製品・フリーウェアも次々と開発・公開・販売が続いている.ここでは,そのダイナミックな熱い現状も詳細に解説されている.
また,それぞれの装置・方法の持ち味は,個々のタンパク質・複合体の解析例においてさまざまに発揮されている.その使い方から最新の成果までが,本分野を代表する研究者達によって解説されている.光岡・横山の稿ではV-ATPaseの動きの構造決定が解説され,岩崎の稿では単粒子解析法が開発当初から適用されてきたウイルスとさらに膜タンパク質の解析が論説されている.横山の稿では単粒子解析の歴史でもあるリボソーム複合体の解析史が最新の成果と共ともに,仁田の稿と成田の稿では細胞骨格の代表である微小管・アクチンとそのレール上を走るモータータンパク質の機構などの解析が,さらに米倉・眞木の稿ではイオンポンプとチャネルの解析について紹介されている.また,石川の稿では微小管とダイニンが組合わさった装置である真核細胞の運動性繊毛構造が詳述され,巨大ウイルスの驚きの構造は岡本・村田の稿で解説され,両章でクライオトモグラフィー法についても詳しく紹介されている.
2クライオ電子顕微鏡を用いた単粒子解析法の原理
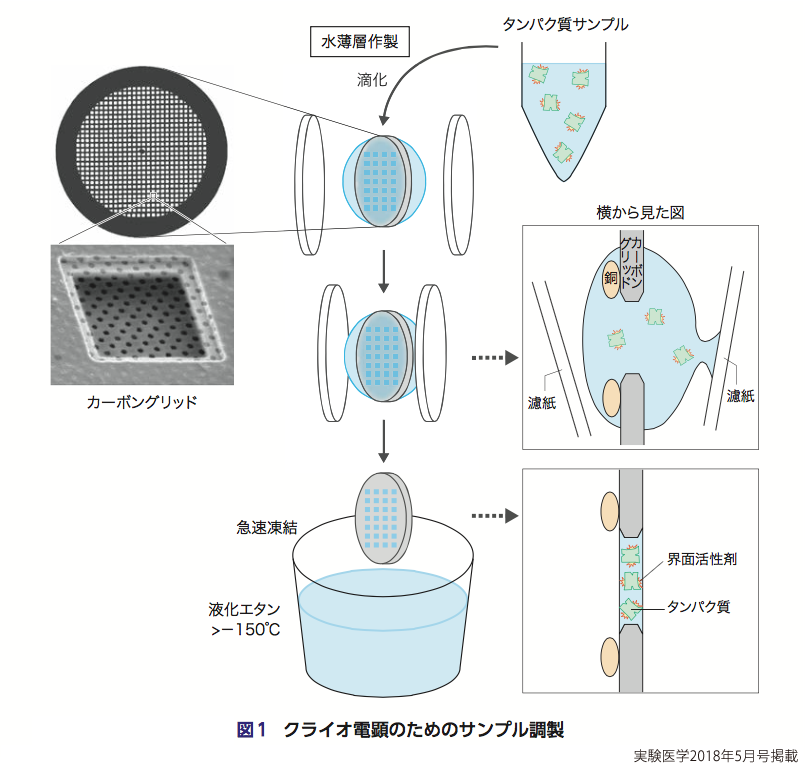
クライオ電顕法では,穴が多く空いたカーボン薄膜を張ったグリッド上に,水に溶けた精製タンパク質を滴下する(図1).次に,両側から濾紙で吸いとり水膜を薄くして※1,膜が弾けてなくなる前に液化エタン中に射出し,急速凍結する.この方法を開発したのがJ. Dubochet教授※2で,水膜を熱電導がよい液化エタン中に投入し急速凍結することで,氷の結晶化を避けvitrified ice(ガラス状氷)が形成されることを発見した.そのなかにタンパク質粒子を閉じ込め(図1),さらに低温のままで霜を付けないようにクライオ電顕に移し(クライオトランスファー),さまざまな方向を向いた粒子を電顕で撮影する(図2).
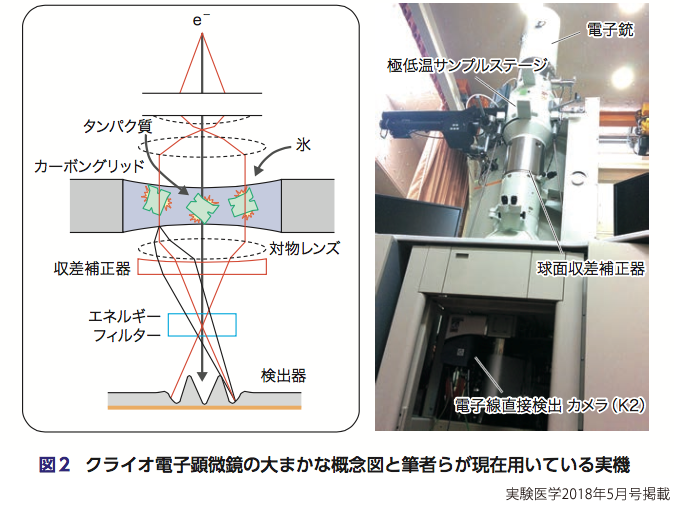
従来型CCDカメラによる撮影では,電子を光に変えてから再び電気シグナルに変える.そのため,変換コストや光学系での拡散によるボケが生じ量子検出効率(detective quantum efficiency,DQE)が低く粒子像がノイズにまみれてしまう点と,秒近くの露出中に粒子がステージの振動などのさまざまな要因によりブレることが問題であった.しかし,これら高分解能への障害は,近年開発された電子線直接検出器(direct detection camera,DDC)の導入により解決された.DDCは,cMOS(complementary metal–oxide–semiconductor)を用いて開発され,途中光を介さないため高感度でDQEが高い.時間分解能も高く,時間ごとに細かく分けて(例えば0.2秒ずつ5秒間:計25枚)粒子を撮影でき,粒子像のズレを補正してから重ね合わせることで鮮明な画像を得ることができる.近年は撮影の自動化も進んできている.
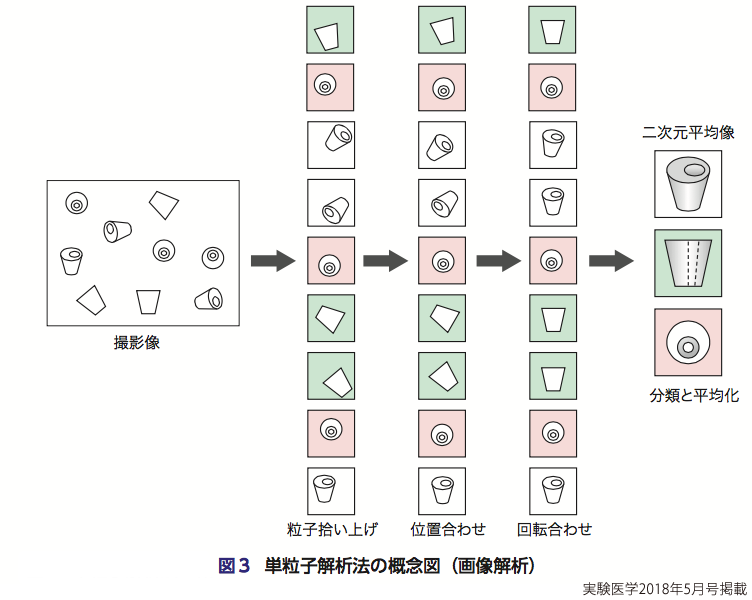
次に得られた元画像から三次元構造を計算する.この三次元再構成のアルゴリズムの道筋は,主にJ. Frank教授※2らのグループにより確立され1),理論的な裏付けの多くはR. Henderson教授※2らによりなされた2).最初,数万を超えるタンパク質粒子像を抽出しライブラリーを形成する(図3).次に画像を平均しより鮮明にするために,これら投影像を,回転位置を合わせた後で相互の類似性から分類する.似た粒子(似た向きの粒子像)同士を重ね合わせ,ノイズの少ない平均像を構築する.次に三次元に構成するために,それぞれの平均像を低分解能の初期三次元構造と照らし合わせてその三次元角(Euler角)を決定し,フーリエ空間を利用して三次元構造を再構成する(図4).この三次元構造からスタートして投影像を作製し,これらを基準像(references)としてライブラリーの粒子像の中心と回転を合わせる.そこから分類によって,よりよい平均化像を作成し,それらのEuler角を求め三次元構造を再構築する.つまり,図3と図4に示したプロセスをくり返す.
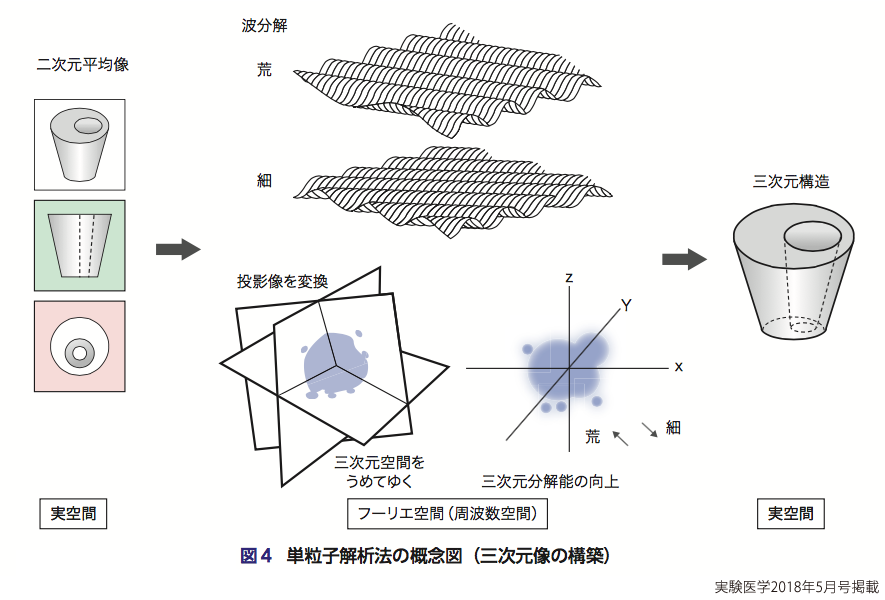
このようなくり返し計算により最適構造に達する.詳しくは安永・荒牧の稿を参照いただきたいが,近年特に画像拾い上げ,分類,三次元再構成などのアルゴリズムや全体の組合わせは,計算精度を上げ計算量を減らすために,ベイズ推定Neural Net,Simulated Annealingなどの人工知能的な手法もとり入れながら大きく進化してきている3)〜5).これらハードソフト両面での方法開発の進展が,単粒子解析法が近年原子分解能へ到達可能になる背景となった.
3解析ソフト
入手できるアルゴリズムとしては,Scheresらによって開発されたRELIONがユーザーフレンドリーでGUI(グラフィカルユーザーインターフェイス)化も進み無料であるため現在最も普及している4).このソフトを用いても単粒子解析法の計算量はやはり多く,現状では例えば,PCにNVIDIA社のGPUカードを数枚揃えGPU計算する必要がある.一般的には,さらにこのようなPCを複数揃える必要がある.他にも,Spider・XMIPP・EMAN・Imagic・Eosなどは,関数が用意されており基本的には自分でスクリプトにより計算を組むスタイルなのでRELIONの標準から離れた計算にも用いられる.これらのソフトは,安定な並列計算のために現状ではUnixがOSとして必須である.
4クライオ電顕単粒子解析法による原子分解能での解析
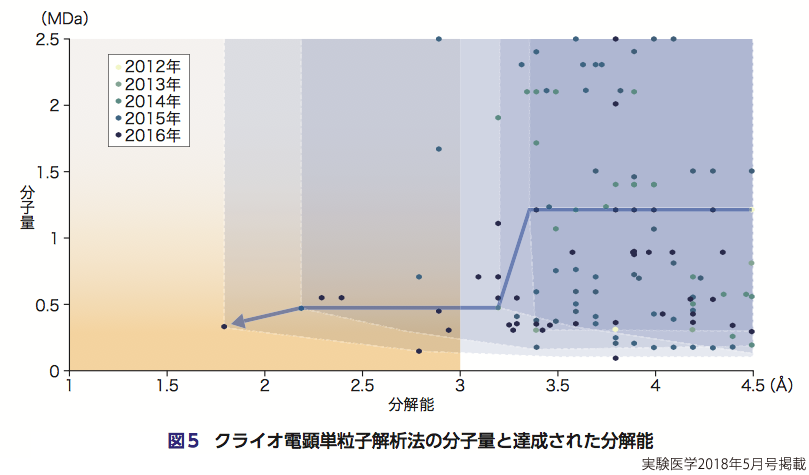
クライオ電顕単粒子法による最初の原子分解能構造は,ChengらのグループによってTRPV1チャネルでNature誌に報告された6).さらに,この号には,続き論文として計3種のリガンドと結合したTRPV1の構造も報告している7).これは,単粒子解析法が薬剤結合構造の決定に適することを反映している.薬剤結合により構造変化したタンパク質を撮影した場合でも,構造変化が極端に大きくなければ,非結合構造を初期構造として使用できることが多い.そこからスタートして,くり返し計算により薬剤結合構造へと到達する.このような過程での単粒子解析法の大きな魅力として,X線結晶解析でのソーキング法※3のように薬剤の浸透による結晶溶けや割れを心配する必要がない点があげられる.現状の単粒子解析の創薬応用における欠点は分解能不足である.現在誰もが認める最高分解能は,465 kDaのβ-ガラクトシダーゼ 8)と334 kDaのグルタミン酸デヒドロゲナーゼ(GDH)9)の結果である.GDHでは分解能が2Åを切りはじめていると思われる.さらにこれを1Å台半ばにする必要がある.この値になるとベンゼン環などがよく見え,薬剤の結合ポケット構造の詳細な記述が可能になると思われる.高分解能化は,物性分野ではすでに幾原らにより0.405Å分解能に達している10).これはSTEM(走査透過電子顕微鏡)を使った結果ではあるが,このような物性分野での高分解能化に大きく貢献したのは収差補正器(Cs-corrector)であり,生物分野でもリボソームの単粒子解析法で使われはじめている11).収差補正器の活用と適したパラメーターの決定は,単粒子解析法の高分解能化において今後焦点の一つになると思われる.また,電顕撮影の自動化は,きわめて重要である.単粒子解析法の最高分解能の一つβ-ガラクトシダーゼでは手動で撮影されたものだが,自動撮影は図5における近年の解析結果の増加と分解能の向上に大きく貢献している.今後,自動化のさらなる進歩が期待されている.
また,単粒子解析法で決定できる分子量の限界は下がってきている(図5).可溶性タンパク質では,分子量65 kDaのヘモグロビンが分解能3.2Åで解析された12).また,膜タンパク質で小さいものは170 kDaのγ-セクレターゼであり3.4Å分解能で解明された13).GPCRでも解析例が出てきており,クラスBに属するカルシトニン受容体がペプチドリガンドとGタンパク質を結合した状態で解析された14).さらに小さなペプチド構造でも,コアタンパク質土台と融合した形で発現させ,土台どうしが対称形に結合することで,分子量を増やし構造解析しやすくする系が提案されている15).一般に,膜タンパク質の解析において,膜貫通部位をとり囲む脂質/界面活性剤(detergent)を工夫することが解析の大きな鍵となる.Chenらのグループは,TRPV1をとり囲むdetergentをlipid Nanodisc(ミセル化せず,膜タンパク質の周囲にのみ脂質二重膜がディスク状に結合する技術)に置換することでより自然なリン脂質構造に近づけることで3.0Åでリガンド結合構造を示した16).
5クライオ電顕の実際―つまづきやすいポイントと解析のコツ
これまでクライオ電顕の原理と最新の解析例を紹介してきたが,クライオ電顕法は,精製したタンパク質を撮影すれば必ず構造を決定できる魔法の箱ではない.つまずきやすいポイントをいくつか紹介したい.

溶液内のタンパク質分子や複合体は,互いに結合していない単分散状態である必要がある.単分散状態であっても,含まれる立体構造が多様なら構造決定は難しい.そのため,写っているタンパク質がどのような状態かの見極めが必要である.結晶成長などの指標がない分,クライオ撮影の前に明確な判断基準がないという意味でかえって難しい面もある.
タンパク質の現状を知るためには,生化学的な方法以外にも視覚的なチェックも役立つ.視覚的なチェックにはクライオ電顕は用いずにより簡便なTEM(透過電子顕微鏡)を用いることが多い.それらによって,タンパク質の精製度の評価と,さらに構造が均一そうかを推定する.より低加速で手軽なスクリーニング用クライオTEM開発という動きもあるが,現状では,小さなタンパク質もはっきりと見やすい負染色法(図6)と簡便な100 kVほどのTEMの組合わせに分がある.例えば99 kDaのイオンチャネルの構造も負染色法で解析されており17),比較的小さなタンパク質にも適用できる.
タンパク質精製では,精製ステップの最後にゲル濾過を入れるのがコツである.タンパク質構造の多型をある程度絞り込むことが期待でき,さらにピークの形やピークシフトによってタンパク質凝集などの情報が得られる.このような情報からも,膜タンパク質の精製条件は検討できる.一般に,detergentや塩濃度・グリセロールなどの保護剤等の条件が精製での構造の安定に重要である.
精製タンパク質粒子を閉じ込める氷の厚さは,見える分子のサイズと分解能にとって重要である.濾紙での水分の吸いとりに要する時間は,タンパク質濃度や粘度の上昇によっても増える.厚さ調節の精度はシステム化などにより向上しているが,装置改良・吸いとり条件のデータベース化などによる一層の改良が求められる.
単粒子解析法では,構造が解けても,多くはコア部分に限られる.例えばTRPV1でも,全体の4割弱の構造は単粒子解析法で決定されていない.それは,タンパク質粒子が結晶化されていないため,周囲が水によるブラウン運動で揺らぎやすいからで,特にこの傾向は粒子の外縁近くで顕著である.分解能をさらに上げ,高分解能領域を拡げるためには,これまでの生理・生化学的な知見の蓄積の活用が重要であり,アミノ酸配列の欠失・置換やリガンド結合によりタンパク質を硬くするアプローチも重要である.この辺りは,結晶解析と同様である.全般的には,日本であまりさかんでなくなってきているタンパク質精製の生化学に立ち返る必要がある.
6多状態からの構造決定
一方,混在する構造が2状態などシンプルな場合は,構造が混在することを逆手にとって,両方の構造を同時に解く方法も存在する.例えば,われわれはGNG(growing neural gas)ニューラルネットによる強力な分類能力を使って,Mg23チャネルの多型が混在した試料より2構造を同時に決定した18).また,タンパク質翻訳において分泌タンパク質などの膜透過を助ける140 kDaのSecDFは,負染色後に個々の粒子をSTEM HAADF(high-argle annular dark field)電顕トモグラフィーにより撮影することで,粒子1個ごとに三次元構造を決定し,三次元構造ライブラリーが作製された.分類して,似た構造を三次元で平均化することにより,クレーンのような分子の動きが示された19).しかし,負染色では粒子乾燥による変形もありあまり分解能は期待できないため,クライオ法でのさらなる構造の精密化が期待される.また,V-ATPaseはATPの加水分解と共役し,分子の軸となる部分を回転させることによってH+を膜を隔てて輸送するポンプであるが,撮影された多状態の粒子を三次元構成の段階で複数に分類することで,回転に伴う複数構造の決定に成功している20)21).将来的には大きく動いた構造が混ざった溶液からでも,タンパク質の三次元的な動きを動画によってあらわせる時代がくるかもしれない.
7クライオ電顕システム導入における注意点
高価なクライオ電顕と氷薄層作製装置および重い計算を可能にするPC設備の導入が必要である.負染色用に,加速電圧100 kv程度の簡易なTEMも必要である.クライオ電顕の仕様を決定する際に重要なポイントがある.それは,機能を盛り込みすぎないことである.盛り込みすぎると,単一機能の実現すら難しくなりやすい.TEMには,膨大な種類のオプションがあり,完全に同じ仕様の電顕にはなかなかお目にかかれない.例えば,真空ポンプだけとっても選択肢がある.その維持のためには,ユーザーの労力も含めさまざまなコストが発生する.幅広い情報収集が何より重要である.クライオ電顕は,導入した時点がシステムアップの終わりではない.場合によってははじまりに近く,運転後に現状での問題点を把握して,電顕メーカーと粘り強く現状評価と改良を行ってゆくことも必要である.
また,システム導入には必須な環境条件がある.クライオ電顕の設置環境に関しては振動が最大の注意点である.対策としては,除振台のうえに電顕を載せる方式や,地面の振動の逆に動いてくれるactive suspensionを土台として導入する手などが考えられる.しかし,一般に低周波振動の消去は簡単ではない.特に,海岸や地下鉄などが近すぎるのは要注意である.磁場変動に関しては,強磁場を変動させる実験装置やエレベーターのモーターなどは電子ビームを曲げる可能性があり,電顕との距離が重要である.距離が大きくなるほど急激に減衰する.音と風も重要である.高分解能クライオ電顕の大きな鏡筒は人間の声などで振動しやすい.部屋によっては防音工事などが必要である.この理由から,ロータリー真空ポンプやエアーコンプレッサー・DDCの制御PCなどは隣室に離して収める必要があることも多い.以上のような対策を構築するために,事前に精密な環境測定が必要である.さらに,近年では高度に環境配慮したTEM室を,TEMとセットで設営するなどの方法が採られることも増えてきている.一般に,TEMの性能が上がれば上がるほど,TEM性能と同じくらい設置部屋の環境対策が重要である.
おわりに
タンパク質以外の生体分子の単粒子解析法もすでにはじまっている.実際,これまでの膜タンパク質の解析でも,結合する脂質も糖鎖も低分解能では見えていた.例えば,Kobilkaらのグループは,GPCRの結晶化のために,精製タンパク質を電顕によって観察することで,脂質が(主に可溶化剤のミセル)タンパク質をおおっていたために,ペプチド配列内にT4リゾチームを導入し突起が安定に細胞外ドメインから突き出す条件を単粒子解析法でスクリーニングした22).それによりユニット間での結合が可能になり,結晶が改善されX線解析により原子分解能へと到達した.また,糖鎖の構造解析もはじまっている.糖鎖は,生体におけるシグナリングと非自己認識などに重要であるが,分子ごとに構造のばらつきが大きく,さらにブラウン運動により揺らぎやすい.しかし,LeeらはHIV1エンベロープ糖タンパク質三量体に,特異抗体のFab断片を2分子結合させ,糖鎖揺らぎを安定化することによりのコア部分の構造を決定した23).今後クライオ電顕は,生体分子だけでなく,ゲルやポリマーなど液体から出すと性質が変わる高分子を中心にさまざまな分子・複合体に広く応用されると思われる.この方法がより広く基礎研究・創薬研究などに使われるように研究・開発を続けていきたい.
謝辞
本研究開発は,新学術領域「構造細胞生物学」,新学術領域「スパースモデリング」,CREST「ピロリ菌の感染と発がんに関する分子機構」,文科省 科研費 萌芽研究(15K14499),産総研 戦略予算 NISPの支援を受けて行われました.
文献
- 「Three-Dimensional Electron Microscopy of Macromolecular Assemblies: Visualization of Biological Molecules in Their Native State」(Joachim Frank), Oxford University Press, 2006
- Rosenthal PB, et al:J Mol Biol, 333:721-745, 2003
- Sorzano CO, et al:J Struct Biol, 148:194-204, 2004
- Scheres SH:J Struct Biol, 180:519-530, 2012
- Ogura T & Sato C:J Struct Biol, 156:371-386, 2006
- Liao M, et al:Nature, 504:107-112, 2013
- Cao E, et al:Nature, 504:113-118, 2013
- Bartesaghi A, et al:Science, 348:1147-1151, 2015
- Borgnia MJ, et al:Mol Pharmacol, 89:645-651, 2016
- Morishita S, et al:Microscopy (Oxf), 67:46-50, 2018
- Fischer N, et al:Nature, 520:567-570, 201
- Khoshouei M, et al:Nat Commun, 8:16099, 2017
- Bai XC, et al:Nature, 525:212-217, 2015
- Liang YL, et al:Nature, 546:118-123, 2017
- Liu Y, et al:bioRxiv:212233, 2017
- Gao Y, et al:Nature, 534:347-351, 2016
- Yazawa M, et al:Nature, 448:78-82, 2007
- Venturi E, et al:Biochemistry, 50:2623-2632, 2011
- Mio K, et al:J Struct Funct Genomics, 15:107-115, 2014
- Nakanishi A, et al:Nat Commun, 9:89, 2018
- Zhao J, et al:Nature, 521:241-245, 2015
- Westfield GH, et al:Proc Natl Acad Sci U S A, 108:16086-16091, 2011
- Lee JH, et al:Science, 351:1043-1048, 2016
著者プロフィール
佐藤主税:1989年東北大学大学院理学研究科生物学専攻博士課程卒業,理学博士.同年に工業技術院電子技術総合研究所に入所.’97年よりグループリーダー.途中,組織が産業技術総合研究所に改組.イオンチャンネルの構造が見たいという動機からクライオ電顕による単粒子解析をはじめる.やがて,この研究方法の開発自体にはまり,スイス,バーゼル大での在外研究を経て現職.常に生物進化の解明をめざしながら研究・開発を進めたい.大学院生・スタッフ募集中.
 人的資源の必要性
人的資源の必要性